10-K: Annual report [Section 13 and 15(d), not S-K Item 405]
Published on May 4, 2026
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For the fiscal year ended
For the transition period from to
Commission File Number:
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) | |
| | ||
| (Address of principal executive offices) | (Zip Code) |
(Registrant’s telephone number, including area code)
Not applicable
(Former name, former address and former fiscal year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required
to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1)
has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months
(or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements
for the past 90 days.
Indicate by check mark whether the registrant has
submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (Section 232.405
of this chapter) during the preceding 12 months (or such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| ☒ | Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check mark
if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards
provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant has
filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting
under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its
audit report.
If securities are registered pursuant to Section 12(b)
of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of
an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ☐
No
As of June 30, 2025 the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the registrant’s common stock outstanding, other than securities held by persons who may be deemed affiliates of the registrant, computed by reference to the closing sales price of $1.76 on June 30, 2025, for the registrant’s common stock, trading on such date, as reported on NYSE American LLC, was approximately $
As
of April 30 2026, there were
DOCUMENTS INCORPORATED BY REFERENCE
TABLE OF CONTENTS
i
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
Various statements in this Annual Report on Form 10-K (the “Annual Report”) of Apimeds Pharmaceuticals US, Inc. are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements involve substantial risks and uncertainties. All statements, other than statements of historical facts, included in this report, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management are forward-looking statements. These statements are subject to risks and uncertainties (some of which are beyond our control) and are based on information currently available to our management. Words such as “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “contemplates,” “predict,” “project,” “target,” “likely,” “potential,” “continue,” “ongoing,” “will,” “would,” “should,” “could,” or the negative of these terms and similar expressions or words, identify forward-looking statements. The events and circumstances reflected in our forward-looking statements may not occur and actual results could differ materially from those projected in our forward-looking statements. Such forward-looking statements are based on current expectations and involve inherent risks and uncertainties, including risks and uncertainties that could delay, divert or change these expectations, and could cause actual results to differ materially from those projected in these forward-looking statements. These risks and uncertainties include, but are not limited to, those factors described under Part I, Item 1A: “Risk Factors.” Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements.
This Annual Report contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this report is generally reliable, such information is inherently imprecise and subject to change.
All written and oral forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We caution investors not to rely on the forward-looking statements we make or that are made on our behalf as predictions of future events. We undertake no obligation and specifically decline any obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws.
We encourage you to read the management’s discussion and analysis of our financial condition and results of operations and our financial statements contained in this Annual Report. There can be no assurance that we will in fact achieve the actual results or developments we anticipate or, even if we do substantially realize them, that they will have the expected consequences to, or effects on, us. Therefore, we can give no assurances that we will achieve the outcomes stated in those forward-looking statements, projections and estimates.
ii
PART I
Item 1. Business
Overview
On December 1, 2025, Apimeds Pharmaceuticals US, Inc., a Delaware corporation (“APUS”, the “Company,” “we,” “us,” or “our”) entered into and closed an Agreement and Plan of Merger (the “Merger Agreement”), with Apimeds Merger Sub, Inc., a Delaware corporation (“Merger Sub”), MindWave Innovations Inc, a Delaware corporation (“MindWave”), Lokahi Therapeutics, Inc., a Nevada corporation (“Bio Sub”), and Erik Emerson, solely in his capacity as representative for the Bio Business (the “Bio Business Representative”). The transactions contemplated by the Merger Agreement are referred to herein as the “Transactions” and the closing of the Transactions is referred to herein as the “Closing”.
Pursuant to the terms and conditions of the Merger Agreement, immediately prior to the Closing, a certificate of merger (the “Certificate of Merger”) was filed with the Secretary of State of the State of Delaware (the “DE SOS”) (such time of the filing of the Certificate of Merger, the “Effective Time”), in accordance with the DGCL. Pursuant to the Certificate of Merger, Merger Sub was merged with and into MindWave (the “Merger”), with MindWave surviving the Merger as the surviving corporation (the “Surviving Corporation”). As a result of the Merger, MindWave became a direct wholly owned subsidiary of the Company. At the Effective Time, all of the property, rights, privileges, powers and franchises of MindWave and Merger Sub vested in the Surviving Corporation and all of the debts, liabilities and duties of MindWave and Merger Sub became the debts, liabilities and duties of the Surviving Corporation. The Closing occurred simultaneously with the execution and delivery of the Merger Agreement on the Closing Date.
At the Effective Time, (i) each share of MindWave common stock issued and outstanding immediately prior to the Effective Time shall be canceled and converted into the right to receive a portion of the Merger Consideration, consisting of shares of Company Preferred Stock; and (ii) each holder of such shares shall receive, for each share of MindWave Common Stock held immediately prior to the Effective Time, a pro rata portion of the Merger Consideration, allocated as follows: (A) a number of duly authorized, validly issued, fully paid and nonassessable shares of Company Common Stock, such that the aggregate number of shares of Company Common Stock issued to all holders of MindWave Common Stock shall equal 0% of the total number of shares of Company Common Stock issued and outstanding as of the date of the Merger Agreement (the “Common Stock Cap”), with each holder’s allocation rounded down to the nearest whole share; and (B) a number of duly authorized, validly issued, fully paid and nonassessable shares of Company Preferred Stock, such that, immediately following the Effective Time, the holders of MindWave Common Stock collectively hold, on an as-converted to Company Common Stock basis, 90.9% of the total issued and outstanding equity securities of the Company (exclusive of the Company Common Stock issued pursuant to clause (A) and calculated on a fully diluted basis). The shares of Company Common Stock and Company Preferred Stock issued to holders of MindWave Common Stock pursuant to the Merger Agreement, on an as-converted and fully diluted basis, shall collectively represent 90.9% of the equity capital of the Company as of the Closing. For purposes of the Merger Agreement, the shares of Company Common Stock, Company Preferred Stock, and MindWave Common Stock issued pursuant to the Merger Agreement are collectively referred to as the “Merger Consideration.”
In connection with the Merger Agreement, on December 1, 2025, certain stockholders of the Company, collectively holding approximately 51% of the Company’s shares of common stock, par value $0.01 per share (the “Common Stock”), approved by written consent in lieu of a special meeting (the “Written Consent”), in accordance with Section 228 of the Delaware General Corporation Law (the “DGCL”) and the Company’s Amended and Restated Certificate of Incorporation, the following actions (the “Corporate Actions”):
| 1. | The Preferred Stock Conversion and Issuance: the issuance of Company Common Stock upon the conversion (the “Preferred Stock Conversion”) of the Series A Convertible Preferred Stock, par value $0.01 per share of the Company (the “Preferred Stock”); |
| 2. | The Notes Conversion and Issuance: the issuance of Company Common Stock upon the conversion of the convertible notes (the “Notes Conversion”) issued by the Company pursuant to that certain Securities Purchase Agreement, entered into by the Company and certain institutional investors on December 1, 2025, and amended by that certain Amendment No. 1 to Securities Purchase Agreement on December 8, 2025; |
1
| 3. | The Reverse Stock Split: a 1-for-10 reverse stock split (the “Reverse Stock Split”) of the Company’s Common Stock, a change in the par value per share of the Company’s Common Stock from $0.01 to $0.001 (the “Change in Par Value”), and a corresponding amendment (the “Charter Amendment”) to the Company’s Amended and Restated Certificate of Incorporation (the “Charter”) to (i) authorize the Board of Directors to effect the Reverse Stock split and (ii) the Change in Par Value; |
| 4. | The 2024 Plan Share Increase: an amendment to the Apimeds Pharmaceuticals US, Inc. 2024 Equity Incentive Plan (the “2024 Plan”) to increase the number of shares of Common Stock issuable under the 2024 Plan to 2,096,679; and |
| 5. | The 2025 Equity Plan: the approval and adoption the Apimeds Pharmaceuticals US, Inc. 2025 Equity Incentive Plan (the “2025 Plan”). |
In connection with such Corporate Actions, the Company filed and mailed an information statement (the “Information Statement”) to its stockholders pursuant to Rule 14c-2 under the Securities Act of 1934, as amended. The Corporate Actions will not become effective until at least 20 calendar days after the mailing of the definitive Information Statement (the “Waiting Period”). Following the expiration of the Waiting Period, or March 25, 2026, the Company expects to file the Charter Amendment with the Secretary of State of the State of Delaware to effect the Reverse Stock Split and the Change in Par Value.
As a result of the Merger, the Preferred Stock Conversion, and the Notes Conversion, a change in control of the Company will occur within the meaning of the NYSE American Company Guide. Accordingly, the Company will be required to submit a new listing application to NYSE American (the “Listing Application”). Although stockholder approval of the Preferred Stock Conversion and the Notes Conversion has been obtained by the Written Consent, which will become effective at the Action Effective Time, the Company does not intend to effect the Preferred Stock Conversion and the Notes Conversion unless and until NYSE American has approved the Listing Application. There can be no assurance that NYSE American will approve the Listing Application. If such approval is not obtained, the Preferred Stock Conversion and the Notes Conversion will not be completed.
INFORMATION ABOUT THE BUSINESS OF APIMEDS PHARMACEUTICALS US, INC.
Unless the context otherwise indicates or requires, all references in this section to “we,” “us,” “our,” “our company” “the Company” and “Apimeds US” refer to Apimeds Pharmaceuticals US, Inc.
The Company conducts its business through two wholly-owned operating subsidiaries, Lokahi Therapeutics, Inc.and MindWave Innovations Inc.
INFORMATION ABOUT THE BUSINESS OF LOKAHI THERAPEUTICS, INC.
Unless the context otherwise indicates or requires, all references in this section to “we,” “us,” “our,” “our company” “the Company” and “Lokahi” refer to Lokahi Therapeutics, Inc.
We are a clinical stage biopharmaceutical company in the process of developing Apitox, an intradermally administered bee venom-based toxin. Our focus is primarily on developing innovative therapies that address inflammation and pain management symptoms associated with knee OA and, to a lesser extent, MS. Apitox is currently marketed and sold by Apimeds Inc. (“Apimeds Korea”) in South Korea as “Apitoxin” for the treatment of OA. Apimeds US is not associated with the market, sale and revenues generated from Apitoxin in South Korea, and Apitoxin has not yet been approved by the FDA for any indication.
Apitox is a purified, pharmaceutical grade venom (bee venom), of the Apis mellifera, or western honeybee, which is classified by the FDA as an active pharmaceutical ingredient (“API”). Bee venom has been used in Asia and Europe to treat pain for hundreds of years. While not FDA approved in a controlled, prescription based biologic environment for defined indications, the use of bee venom has been FDA approved as a “under the skin injection” to reduce the allergic reactions to bee stings. Apimeds Korea has developed a proprietary method and process for turning extracted bee venom into a lyophilized powder for reconstitution prior to intradermal dose injections, which they sell in South Korea as Apitoxin. We intend to use a similar process with respect to Apitox, pursuant to the Business Agreement, which gives us a license to utilize all prior clinical development data associated with Apitoxin. The advancement of extracted bee venom for treatment of inflammatory conditions, including but not limited to knee OA and MS is speculative but based on direction provided by prior clinical data.
2
Apimeds Korea successfully completed Phase I, Phase II, and Phase III trials in OA in 2003, at which point Apitoxin was approved by the Korean Ministry of Food and Drug Safety (“MFDA”) to treat pain and mobility in patients with OA. Since 2003, a post-marketing/approval safety study in South Korea followed 3,194 patients from 2003 through 2009, with no serious adverse events. The purpose of a Phase I trial is to test to determine whether a new treatment is safe and look for the best way to give the treatment. Phase II trials test to determine whether a condition or disease responds to the new treatment. Phase III trials test to determine whether a new treatment is better than a standard treatment.
In 2013, the first of two required U.S. Phase III clinical trials was authorized to enroll patients to study the use of Apitoxin to study the same indication as approved in South Korea in 2023 — treatment of pain and lack of mobility in patients with OA (the “Apimeds Korea Phase III OA Trial”). The Apimeds Korea Phase III OA Trial (330 patients) was completed in 2018, and displayed no serious adverse events.
Based on the results from the Apimeds Korea Phase III OA Trial, which demonstrated therapeutic (statistical and clinically significant improvements in all outcome measures of pain, physical function, and disease assessment) effect compared to the placebo group, but in combination with prior development by Apimeds Korea, did not meet the FDA’s standards for approval, as the study population was too small and the methods for handling missing data were inadequate, resulting in a study that did not demonstrate a significant treatment effect. We will be pursuing a second Phase III trial to meet agreed upon FDA standards. Based on results from the Apimeds Korea Phase III OA Trial, we have evaluated the most appropriate population, defined as advanced knee OA patients, which will range from defined grade 2, 3 and 4 within this treatment group, to continue to progress our own Phase III trial. Pursuant to our previous correspondence with the FDA, we have designed and will implement our Phase III trial to best address our patient population, appropriate dosing, and the most effective way to evaluate Apitox in meeting the patient population’s needs.
We believe the progress we are making in clinical trials provides us support in our belief in the potential of Apitox to be an innovative therapy. We aim to treat the inflammation and pain management symptoms associated with knee OA and to help manage the devastating symptoms of this disease. In the future, we also aim to leverage our research in knee OA to investigate how Apitox may be used to treat similar symptoms associated with MS.
Treatment of OA
OA is typically treated with painkillers known as non-steroidal anti-inflammatory drugs (NSAIDs). These medications have an anti-inflammatory and pain-relieving effect. These medications include ibuprofen (Motrin, Advil) naproxen (Aleve) and diclofenac (Voltaren and others). All of these medications work by blocking enzymes that cause pain and swelling. The problem is that some of those enzymes also help blood to clot and protect the lining of your stomach. Without them, you can bruise easily, develop ulcers and may even bleed in your intestines. NSAIDs also increase your chance of heart attack, stroke and heart failure. The risk increases the longer you use them and the more you take. We believe Apitox could be a successful alternative to NSAIDs in the treatment of the inflammation and pain management symptoms associated with OA without the harmful side effects.
According to MedicalNewsToday, OA is the most common form of arthritis, affecting around 500 million people worldwide, or around 7% of the global population. Currently, in the United States, over 32 million people suffer from OA. As the 15th highest cause of years lived with disability (YLDs) worldwide, the burden OA poses to individuals is substantial, characterized by pain, activity limitations, and reduced quality of life. The economic impact of OA, which includes direct and indirect (time) costs, is also substantial, ranging from 1 to 2.5% of gross national product (GNP) in countries with established market economies, like the United States. Though trends in OA prevalence vary by geography, the prevalence of OA is projected to rise in regions with established market economies such as North America and Europe, where populations are aging and the prevalence of obesity is rising.
While OA can occur in any joint, it occurs most frequently in the knee, which, according to ScienceDirect, currently accounts for 365 million cases worldwide and 61% of YLDs lost due to OA, followed by the hand.
Our current efforts are focused on the development of Apitox in the United States for the treatment of inflammation and pain management relating to OA in the knee.
3
Treatment of MS
Additionally, we believe the previous clinical trial success of Apimeds Korea with respect to the use of Apitoxin to treat symptoms associated with knee OA, and pending the success of our anticipated Phase III trial in knee OA, we will be in a position to further explore the use of Apitox as a potential treatment for the symptoms of MS. MS is a chronic disease of the central nervous system. It is an autoimmune condition that is characterized by the body’s own immune cells (macrophages and lymphocytes) attacking the myelin that coats nerve cells, which can lead to inflammation throughout the central nervous system. MS is an unpredictable disease that affects people differently. Some people with MS may have only mild symptoms. Others may lose their ability to see clearly, write, speak, or walk when communication between the brain and other parts of the body becomes disrupted.
MS is the most common progressive neurologic disease of young adults worldwide. A study funded by the National MS Society estimates that nearly one million individuals are currently affected by this disease in the United States. The total economic burden of MS in the United States is estimated to be $85.4 billion, with $63.3 billion in direct medical costs and $22.1 billion in indirect and nonmedical costs. MS typically affects patients at a young age, resulting in a greater loss of productivity and quality of life.
Beta interferon drugs are among the most common medications used to treat MS. Interferons are signaling molecules that regulate immune cells. Potential side effects of these drugs include flu-like symptoms (which usually fade with continued therapy), depression, or elevation of liver enzymes.
Pain from MS can be felt in different parts of the body. Trigeminal neuralgia (facial pain) is treated with anticonvulsant or antispasmodic drugs, or less commonly, painkillers. Central pain, a syndrome caused by damage to the brain and/or spinal cord, can be treated with gabapentin and nortriptyline. Treatments for chronic back or other musculoskeletal pain may include heat, massage, ultrasound, and physical therapy.
OA and the Current Standard of Care
OA is a degenerative joint disease in which the tissues in the joint break down over time. It is the most common type of arthritis and is more common in older people. People with osteoarthritis usually have joint pain and, after rest or inactivity, stiffness for a short period of time.
There are four stages of OA: (1) Minor — minor wear-and-tear in the joints and little to no pain in the affected area, (2) Mild — more noticeable bone spurs, the affected area feels stiff after sedentary periods and patients may need a brace, (3) Moderate — cartilage in the affected area begins to erode, the joint becomes inflamed and causes discomfort during normal activities, and (4) Severe — the patient is in a lot of pain, the cartilage is almost completely gone leading to an inflammatory response from the joint, and overgrowth of bony spurs may cause severe pain.
With the progression of OA of the knee, there is obvious joint inflammation which causes frequent pain when walking, running, squatting, extending or kneeling. Along with joint stiffness after sitting for long or when waking up in the morning, there may be popping or snapping sounds when walking.
The data from the Apimeds Korea Phase III OA Trial suggest that Apitox would have the most potential in treating OA in stages 3 and 4.
MS and the Current Standard of Care
MS is increasingly recognized as a neurodegenerative disease triggered by an inflammatory attack of the central nervous system. There is no cure for multiple sclerosis. Treatment typically focuses on speeding recovery from attacks, reducing new radiographic and clinical relapses, slowing the progression of the disease, and managing MS symptoms.
MS is unpredictable and can vary substantially from person to person. MS is divided into four types: clinically isolated syndrome (CIS), relapsing-remitting MS (RRMS), secondary progressive MS (SPMS) and primary progressive MS (PPMS).
CIS refers to a first episode of neurologic symptoms caused by inflammation and demyelination in the central nervous system.
4
RRMS, the most common disease course, shows clearly defined attacks of new or increasing neurologic symptoms. These attacks are also called relapses or exacerbations. They are followed by periods of partial or complete recovery, or remission. In remissions, all symptoms may disappear or some symptoms may continue and become permanent. However, during those periods, the disease does not seem to progress.
SPMS follows the initial relapsing-remitting course. Some people diagnosed with RRMS eventually go on to have a secondary progressive course, in which neurologic function worsens progressively or disability accumulates over time.
With PPMS, neurologic function worsens or disability accumulates as soon as symptoms appear, without early relapses or remissions. PPMS can be further characterized as either active (with an occasional relapse and/or evidence of new MRI activity over a specified period of time) or not active, as well as with progression (evidence of disability accrual over time, with or without relapse or new MRI activity) or without progression.
Patients with MS tend to be more educated about their disease and better organized than patients with other diseases, resulting in patients that are aggressive in their approach to treatment. This is due to MS impacting otherwise healthy people in the prime of their lives.
MS treatment has undergone significant evolution in the last ten years with the development and approval of certain new drugs, including several oral agents such as Ocrevus, in the United States. These new agents not only give patients additional treatment options, but also have improved the efficacy and safety of treatment for MS overall. In general, these drugs are “disease modifying agents,” intended to slow down the immune mediated damage to the myelin sheaths that underlie symptoms in MS. However, they often do not adequately address the symptoms that MS patients experience such as walking problems, bladder control, dizziness, and especially pain. A 2022 study estimated that the average cost of treatment for patients with MS is approximately $88,000 annually. The out-of-pocket expense for patients can be significantly reduced through certain insurance plans. However, we believe there is the ability for Apitox to be positioned as an important and cost-effective therapy.
We believe the data from the Apimeds Korea Phase III OA Trial suggest that Apitox may have the potential as an adjunctive therapy for all four types of MS. We intend to explore Apitox as a potential adjunctive therapy through non-registered corporate sponsorship studies to begin determining the appropriate MS patient populations.
Market Opportunity
We believe there is a significant market opportunity in the United States for Apitox in the treatment of certain symptoms of knee OA and eventually MS. According to Precedence Research the osteoarthritis therapeutics market size accounted for $8.28 billion in 2022 and it is expected to hit around $20.24 billion by 2032, expanding at a CAGR of 9.4% from 2023 to 2032. Although OA can damage any joint, the disorder most commonly affects joints in your hands, knees, hips and spine. OA symptoms can usually be managed, although the damage to joints can’t be reversed. Apitox has certain anti-inflammatory properties, which we believe give it significant potential to help treat the symptoms of certain chronic diseases that involve difficult to control pain and inflammation.
According to Pharmaceutical Technology the MS market size in the United States accounted for $10.73 billion in 2022 and is expected to hit $24.4 billion by 2030, expanding at a CAGR of 10.32%. Starting in the first quarter of 2025, we intend to begin the early prosecution of appropriate MS patient populations through non-registered corporate sponsorship studies. Subject to FDA approval, our development of Apitox in the United States will in the near term, have two distinct focuses (i) the treatment of the certain symptoms of knee OA and (ii) the quality of life issues surrounding knee OA, such as pain and lack of mobility.
Living with a chronic disease is challenging, as it interferes with physical, mental, and social functions and thus greatly affects a person’s quality of life. Indeed, chronically ill patients are facing major struggles such as higher expenditures, social isolation and loneliness, disabilities, fatigue, pain/discomfort, feelings of distress, anger, hopelessness, frustration, anxiety, and depression. There is the general assumption that symptom reduction increases a patient’s quality of life. Our approach with Apitox centers around this concept — effectively treating certain symptoms of the patient’s disease, thus improving their overall quality of life. Bee venom has been shown to have anti-inflammatory effects. At low doses, bee venom can suppress inflammatory cytokines such as interleukin-6 (IL-6), IL-8, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α). A decrease in the signaling pathways responsible for the activation of inflammatory cytokines, such as nuclear factor-kappa B (NF-κB), extracellular signal-regulated kinases (ERK1/2) and protein kinase Akt, and porphyromonas gingivalis lipopolysaccharide (PgLPS)-treated human keratinocytes has been associated with treatments involving bee venom. We believe the driver of pain in the highest category of OA is correlated to the key inflammatory elements treated by bee venom, meaning the evaluation of our Phase III data may lead to a small indication for narcotic use reduction in the treatment of stage 4 OA. Lokahi partners with universities to give students hands-on exposure to the strategic side of biopharma, from evaluating clinical assets to understanding intellectual property, market dynamics, and go-to-market strategies. The ai² Futures Lab™ functions as both a discovery engine for potential therapeutic assets and a training ground for the next generation of biotech and business leaders.
5
Our Product Candidate
Apitox is purified honeybee (Apis mellifera) venom manufactured as a lyophilized powder for reconstitution in 0.5% preservative-free lidocaine (lmg/mg) prior to intradermal dose injections that are administered up to 1,500 micrograms per weekly visit. The biologically active components include melittin (40-50%), apamin (2-3%), mast cell degranulating (“MCD”) peptide (Peptide 401,2-3%), phospholipase A2 (10-15%), hyaluronidase (1.5-2%) and other components in small amounts, including dopamine and norepinephrine. According to a publication entitled “Pharmacological effects and mechanisms of bee venom and its main components: Recent progress and perspective” by Shi et al., certain components of honeybee venom have been found to have both anti-inflammatory and analgesic effects. The anti-inflammatory and analgesic effects are attributed to the presence of Peptide 401, adolapin and other components that inhibit prostaglandin synthesis. The hormone-stimulating effects are attributed to the presence of melittin, cardiopep and other components that stimulate the pituitary-adrenal axis to produce cortisol. Results from an animal study entitled “Effect of bee venom and melittin on plasma cortisol in the unanesthetized monkey” published by Vick et al., indicate that melittin appears to stimulate the production of cortisol from the adrenal gland. The immune-modulating effects, especially as it pertains to MS, are suggested to be mediated by CD4+CD2S+Foxp3+ regulatory T cells (Tregs) that are influenced by phospholipase A2. While the exact mechanism of action of Apitox is not fully understood, research such as the publication entitled “Therapeutic Use of Bee Venom and Potential Applications in Veterinary Medicine” by Bava et al., suggests that certain components in Apitox may ameliorate immune-inflammatory responses associated with MS. Such studies suggested that treatments with melittin prevent inflammatory cytokine expression and produces anti-inflammatory effects. The proposed indication for Apitox is to provide add-on therapy for the signs and symptoms of MS in patients whose condition is relapsing-remitting (RRMS), primary-progressive (PPMS) or secondary progressive (SPMS).
Clinical Development History
Founded in 1989, Apimeds Korea pursued a traditional drug development process in South Korea for Apis mellifera, the bee venom API for Apitoxin. Apimeds Korea completed a formal preclinical study to validate dosing and safety for human administration with a focus on antigenicity and toxicology in 1993.
A Phase I trial was completed in 1994, studying the toxicity and safety of Apitoxin in 20 healthy subjects. The purpose of the Phase I trial was to determine if therapeutic doses of Apitoxin was safe and to identify possible side-effects, if any. Injections of Apitoxin were given two to three times a week, for a total of 12 sessions spanning over four to six weeks. Laboratory and physical examination of the subjects included (i) serum cortisol levels (to see if Apitoxin stimulated the release of cortisol), (ii) serum ionized calcium level (to determine if Apitoxin decreased the serum calcium level), (iii) urinalysis, (iv) hematology and blood chemistry, and (v) vital signs. The Phase I trial demonstrated that there were no significant changes pre- and post-testing of the serum cortisol levels, serum ionized calcium levels, hematology, blood chemistry, urinalysis, and vital signs after the subjects were injected with Apitoxin according to the protocol. There were no significant physiological changes in the clinical evaluations of the subjects and localized itching was the most frequent side effect and was managed with ice packs or external anti-itching gels. No severe side effects or aftereffects were observed. The Phase I trial indicated that Apitox is safe for humans when applied in therapeutic doses.
6
The Phase I trial was followed by a Phase II trial in 101 subjects to determine the efficacy of Apitoxin at various dose levels. This was a randomized active-controlled clinical trial with three groups receiving the study drug at various dose levels and one group receiving the control drug (nabumetone) for a six-week period. Patients received twice weekly injections of Apitox intradermally at dosages titrated to a maximum of 0.7 mg (Group A), 1.5 mg (Group B), and 2.0 mg (Group C) for a period of six weeks. Control group patients (Group D) received 1,000 mg of nabumetone orally each day for the same six-week period. There were 25, 26, 25 and 25 patients assigned to Groups A, B, C and D, respectively. Efficacy of treatment was evaluated by the physician investigators using a 4-point Likert-like symptom severity rating scale developed by the authors to assess Pain, Disability and Physical Signs. A similar 5-point scale was used for patient self-evaluation. Safety of the Apitoxin injection was evaluated by patient reaction, hematologic examination, and laboratory chemistry analysis of blood and urine. Efficacy data was reported for the 81 patients who completed the study. While there were no significant differences in symptom severity scores among the four groups at baseline, symptom scores were significantly better in the bee venom injection groups than in the control group at six weeks and 10 weeks after the start of treatment (p<0.01). A treatment was considered effective if there was a 20% improvement from baseline in symptom scores after 6 weeks of treatment. Based on this definition, therapy demonstrated overall efficacy in 70.0% of patients in Group A, 85.7% in Group B, 90.0% in Group C, and 61.9% in Group D (drug control). Overall efficacy was significantly greater in treatment Groups B and C combined than in the nabumetone-treated control group D (p<0.0177). Importantly, efficacy of treatment among all patients treated with Apitoxin injection was greater than among nabumetone-treated patients for each category assessed: Pain: 85.2% versus 76.2%; Disability: 77.0% versus 71.4%; and Physical Signs: 62.3% vs. 23.8%. It is also noteworthy that, unlike the drug control group, the Apitoxin injection groups continued to demonstrate improved symptom scores at four weeks after the last treatment (10 weeks). There were no significant changes in vital signs or results of laboratory examinations of any patient in this clinical trial. Localized itching was experienced by all patients who received Apitox injections. Itching at the injection site generally lasted for two to three weeks; several patients had this reaction for a longer period. This Phase II study showed that Apitoxin was significantly more effective than the control drug, nabumetone, in the treatment of knee and spinal osteoarthritis patients. It clearly showed that improvement in pain, disability and physical signs was greater in the bee venom injection groups than in the nabumetone control group. No significant side effects developed at the therapeutic doses studied. However, research should be continued to minimize itching and pain at bee venom injection sites, and possible allergic reaction should always be considered with treatment at high doses.
In 2002, a formal Phase III double-blind, placebo-controlled trial was completed with 407 subjects (311 of which obeyed the trial protocol and completed the clinical study). The purpose of the Phase III trial was conducted to verify the efficacy and safety of the medicine resulting from the prior Phase I and Phase II trials. The therapeutic course treatment included a total of 12 injections over a period of 6 weeks. Final evaluations were completed in the 8th week, following two weeks of no injections. During the trial period, laboratory tests were carried out three times (before injection, in the second week, in the sixth week), and the efficacy evaluation was performed four times (before injection, in the second week, in the sixth week, and in the eighth week). Safety of the Apitoxin injection was evaluated by hematologic examination, measurement of cortisol and calcium levels, and laboratory chemistry analysis of blood and urine. The primary efficacy variable for the trial was the ratio of the subjects who showed more than 20% improvement in the total points of test items for efficacy evaluation 6 weeks after injection, compared with the total points before injection of the medicine (the “improvement rate”). Data obtained from subjects of the clinical test were analyzed by two methods, ITT (Intention to Treat) analysis and PP (Per Protocol) Among 310 subjects who participated in the efficacy evaluation, 153 and 157 patients belonged to the Apitoxin group and the nabumetone group, respectively. For the Apitoxin group, the ratio of the subjects who showed more than 20% improvement in the total points was 48.70% (75/154 subjects, 95% confidence interval (“CI”): 40.8~56.6%), while for the nabumetone group, it was 46.15% (72/156 subjects, 95% CI: 38.3~54.0%), indicating that the improvement rate in the Apitoxin group was greater than in the nabumetone group; however, there was no statistical significance. (p=0.6533). Among a total of 407 subjects (Apitoxin group: 204; Nabumetone group: 203), 38.24% (78/204) of the Apitoxin group showed more than 20% improvement during the 6th week of injection, while 38.42% of the Nabumetone group improved by more than 20%, indicating that the two groups showed similar improvement rate (p=0.9688). The second efficacy variable was the improvement rate during the 8th week (2 weeks after the completion of the final injection). According to results from comparing the total points of efficacy evaluation items during the second week after completion of injection (during the 8th week after injection) with the total points before injection, 58.44% (90/154) of the Apitoxin group showed a higher improvement rate than during the 6th week (48.70%), while 42.95% (67/156) of the Nabumetone group showed lower improvement rate than during the 6th week (46.15%). There was statistical difference in total point of efficacy evaluation items between the two groups (p=0.0064). These results suggest that even after treatment stops, the efficacy of Apitoxin continues. With respect to safety, among a total of 407 subjects who participated in the safety evaluation, 69 (33.82%) of the Apitoxin group showed an adverse event, while 59 (29.06%) of the Nabumetone indicated adverse event. These results indicate that the Apitoxin group had an elevated adverse event rate than the Nabumetone group, but there was no statistically significant difference between the two groups (p=0.3526).
In May 2003, MFDA granted approval for the use of Apitoxin in the treatment of pain and mobility in patients with OA. A post-marketing/approval safety study in South Korea followed 3,194 patients from 2003 through 2009, with no serious adverse events or negative safety signals.
7
In 2013, preliminary Phase III clinical trials were authorized to enroll patients by the FDA to study the same indication approved in South Korea — treatment of pain and lack of mobility in patients with OA. The results of the preliminary Phase III clinical trial indicated statistical and clinically significant improvements in all outcome measures of pain, physical function, and disease assessment in the study group. The study group included 330 patients with diagnosed osteoarthritis of the knee. The subjects were evaluated for relief of pain using Western Ontario and McMaster Osteoarthritis Index (WOMAC) and physician and patient global assessments. The primary efficacy measure was relief of pain and inflammation over a 12-week treatment period after randomization into the trial. The secondary efficacy measure was improvement of mobility. Treatment effect will be compared in a 2-1 Apitox vs active control. Compared with the placebo group (histamine), subjects in the Apitox group who received a maximum dose (1500 micrograms) at each weekly visit over 12 weeks showed a significantly more improvement in all outcome measures (WOMAC pain, WOMAC physical function, visual analog scale (“VAS”) pain, patient and physician global assessments of OA). Further, post hoc analyses showed that a statistically significant greater percentage of Apitox-treated subjects had at least a 40% and 60% reduction in WOMAC pain as compared to placebo-treated subjects. Sensitivity analyses confirmed the validity of the statistical methods and population definitions. The improvements in pain endpoints were highly significant for both the modified intention to treat and per protocol populations and the improvement was sustained during the four weeks following Apitox treatment.
Except for an expected higher incidence of injection site reactions (<5%) in the Apitox group, the overall safety profiles were comparable between the treatment groups. A serious adverse event of the anaphylactic reaction occurred in an Apitox-treated subject because of a quick injection rate. However, the subject was treated, and the event was resolved within one day. The incidence of adverse events overall was similar between the Apitox and Placebo groups (49.0% and 46.3%, respectively), and there were no clinically meaningful changes, within and between groups, in laboratory parameters, vital signs, physical examination, or electrocardiogram results.
During Apimeds Korea meetings with the FDA, the FDA highlighted concerns regarding the opioid crisis. As Apitoxin has been previously approved in South Korea, we believe Apitox could be a viable treatment option within the United States after additional clinical investigation, including our anticipated Phase III trial. Initially, Apimeds Korea elected not to pursue the OA indication in the United States based on its evaluation of potential market adoption and the existing competitive environment for OA. Based on results from the Apimeds Korea Phase III OA Trial and correspondence with the FDA, we believe we are now in a position to continue to advance our Phase III trial for knee OA.
We intend to conduct an additional Phase III trial in knee OA. Based on our previous correspondence with the FDA, we have started to design and will implement our Phase III trial to best address our patient population of patients with grade 2, 3 and 4 knee OA, appropriate dosing, and the most effective way to evaluate Apitox in meeting a patient’s needs. This trial will be an update to the plan of execution based on review of data, discussions with former principal investigators from Apimeds Korea. Upon successful completion and FDA clearance of our Phase III trial in knee OA, we will be positioned to submit a BLA.
We intend that the purpose of this trial will be to evaluate the effectiveness of Apitox in the treatment of grade 2, 3 and 4 OA of the knee. The trial will be designed with a specific focus on the identified subgroup from which we see the highest degree of benefit.
8
The following table summarizes the preliminary clinical trial activity by Apimeds Korea with respect to Apitoxin:
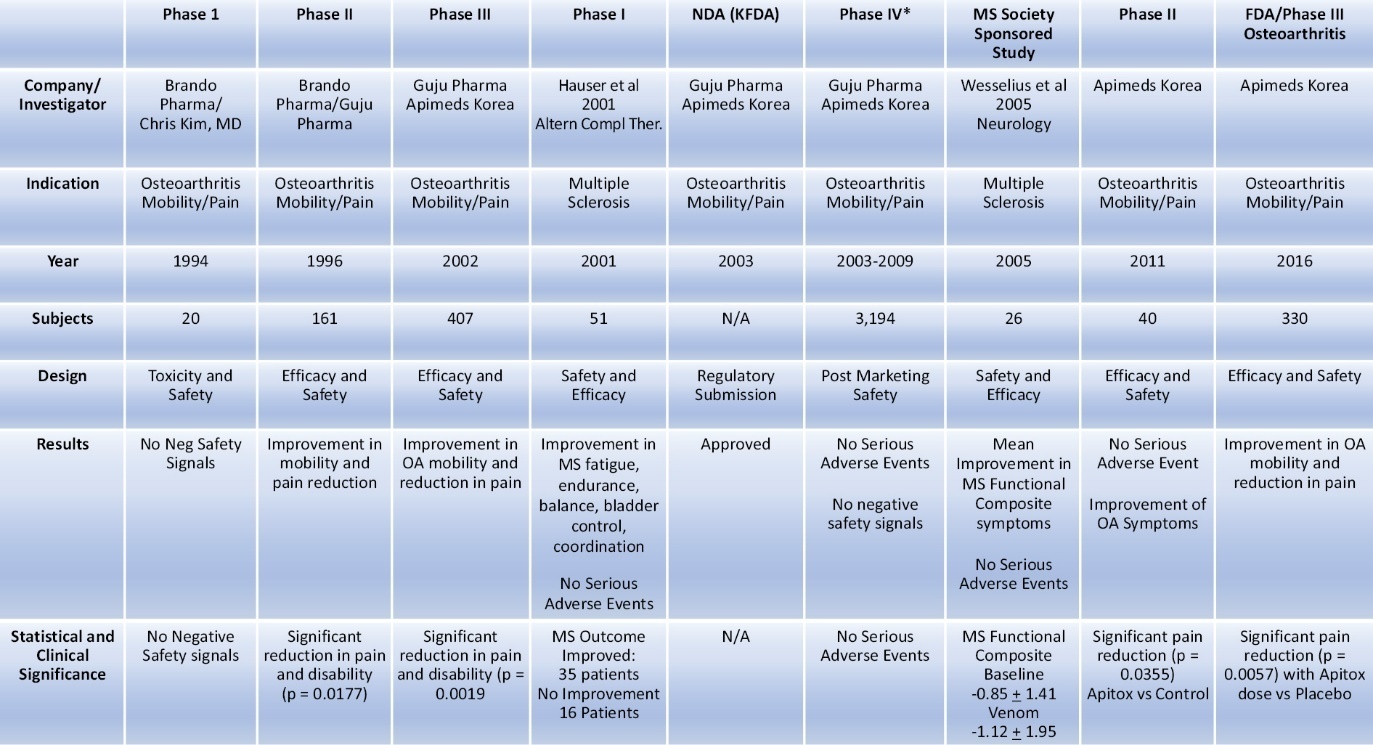
Preliminary Clinical Data in MS Patients
The United States data from the literature on bee venom studies in MS patients, Table A (Hauser et al. 2001) below, showed clinically significant improvements in disability symptoms following treatment.
In Table A, results were categorized into the following groups: dramatic disability improvement (>12 points on the Related Observable Symptom Scale (“ROSS”), good improvement (7-12 points on ROSS), minimal improvement (<7 points on ROSS), no improvement (<2 points on ROSS), and negative (any total negative response on ROSS). Descriptive analysis of the ROSS clinical outcomes showed that more than 68% of MS patients showed some kind of positive improvement in disability (dramatic, good or minimal) and 58% demonstrated a marked improvement (dramatic or good).
Table A. Summary of Patient Disability Improvement to Bee Venom Treatment Using ROSS
| N | % of Participants | Follow-up Survey (% improvement) | Related Observable Symptoms Scale (points improvement) | |||||||
| Dramatic | 15 | 29.4 | % | >30%, or | >12 points | |||||
| Good | 15 | 29.4 | % | 10 – 29%, or | 7 – 12 points | |||||
| Minimal | 5 | 9.8 | % | <10%, or | <7 points | |||||
| None | 15 | 29.4 | % | <2%, or | <2 points | |||||
| Negative | 1 | 2.0 | % | Any total negative response | Any total negative response | |||||
After 1 year of bee-venom injections, 68.6 percent of participants showed improvement. N = number of participants.
Apimeds Korea used data from its first Phase III clinical trial for OA and peer reviewed publications, including those referenced in Table A above and formal Phase I (the “Castro Phase I Trial”) and Phase II (the “Wesselius Phase II Trial”) publications specific to MS, to support its submission in 2014 of its Investigational New Drug Application (“IND”) 122804 (A Phase III, Multi-Center, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study to Evaluate the Safety and Efficacy of Apitox Add-on Therapy for Improving Disability and Quality of Life in Patients with Multiple Sclerosis).
9
Castro Phase I Trial
The Castro Phase I Trial involved a total of nine bee venom nonallergic patients with progressive forms of MS, who were 21 – 55 years of age with no other illnesses. The subjects distributed across four groups (A, B, C, and D) and followed a structured 1-year immunization schedule. Hyperreactivity to bee venom was evaluated by questionnaire, physical examination, and a battery of hematologic, metabolic, and immunologic tests. Responses to therapy were evaluated by questionnaire, functional neurological tests, and changes in measurement of somatosensory-evoked potentials. While no serious adverse allergic reactions were observed in any of the subjects, four experienced worsening of neurological symptoms, requiring their discontinuation in the study. The observed negative effects could not be conclusively attributed to adverse reactions arising from the administered therapy. Of the remaining five subjects, three reported subjective amelioration of symptoms and two exhibited objective improvement. Despite suggesting safety in this preliminary study, the small sample size precluded definitive conclusions regarding the efficacy of the treatment for MS. Larger and more carefully conducted multicenter studies were required to establish efficacy.
Wesselius Phase II Trial
The Wesselius Phase II Trial involved a randomized crossover study of 26 patients diagnosed with relapsing-remitting or relapsing secondary progressive MS. Participants were assigned to 24 weeks of medically supervised bee sting therapy, or a control period of 24 weeks of no treatment. Live bees (up to a maximum of 20) were used to administer bee venom three times per week. The primary outcome was the cumulative number of new gadolinium-enhancing lesions on T1-weighted MRI of the brain. Secondary outcomes were lesion load on T2*-weighted MRI, relapse rate, disability (Expanded Disability Status Scale, Multiple Sclerosis Functional Composite, Guy’s Neurologic Disability Scale), fatigue (Abbreviated Fatigue Questionnaire, Fatigue Impact Scale), and health-related quality of life (Medical Outcomes Study 36-Item Short Form General Health Survey). The results of the Wesselous Phase II Trial indicated that during bee sting therapy, there was no significant reduction in the cumulative number of new gadolinium-enhancing lesions. The T2*-weighted lesion load further progressed, and there was no significant reduction in relapse rate. There was no improvement of disability, fatigue, and quality of life. Bee sting therapy was well tolerated, and there were no serious adverse events. In this trial, treatment with bee venom in patients with relapsing multiple sclerosis did not reduce disease activity, disability, or fatigue and did not improve quality of life measured using gadolinium-enhancing MRI.
From June 2014 to June 2018, Apimeds Korea corresponded with the FDA and there were no clinical holds at that time. Sponsorship of IND 122804 was transferred from Apimeds Korea to us in October 2020. On September 21, 2021, we responded to customary non-clinical hold comments from the FDA. In November 2021, we received a customary clinical hold from the FDA due to the retirement of the former principal investigator. We have subsequently updated the FDA with a new principal investigator via our Chief Medical Officer, Dr. Christopher Kim. In February 2023, the FDA removed the clinical hold and concluded it may be initiated. We have subsequently made the strategic decision to focus our efforts and capital on our Phase III trial in knee OA, and instead focus our MS efforts on the early prosecution of appropriate MS patient populations through non-registered corporate sponsorship studies.
10
Our Commercialization Strategy
We are dedicated to the effective implementation of regulatory, clinical and legal strategies to create value in Apitox. The effective execution of this strategy will provide us the opportunity to evaluate and potentially acquire other assets that fit within our space for development.
Manufacturing
We intend to continue to engage a third-party manufacturer, Piramal Pharma Solutions, in Lexington, Kentucky to support our Phase III trial and, if Apitox is approved by the FDA, commercial manufacturing. This manufacturer has dedicated experience in development and technology transfer of sterile dose formulations, including liquid and lyophilized formulations.
Research and Development
We are currently engaged exclusively in the clinical development of Apitox for continued use in knee OA through a Phase III trial in knee OA and potential use for MS through the early prosecution of appropriate patient populations through non-registered corporate sponsorship studies.
Sales and Marketing
The healthcare providers associated with the treatment of inflammation and pain management symptoms associated with OA and MS are not limited to one specialist but involve a comprehensive team of providers focused on slowing the progression of the disease along with the physical, emotional and day-to-day management of the condition. Each of these providers represents a potential customer for Apitox.
Apitoxin, which will be known as Apitox in the United States, has established technological credibility through its preclinical testing, Phase I, Phase II and preliminary Phase III clinical studies completed by Apimeds Korea. Apimeds Korea received regulatory approval for Apitoxin by the MFDA in South Korea, as well as long-term safety data from treatment of patients in Korea from 2003 to 2009. There were no serious adverse events from over 3,000 patients monitored, and Apitoxin has been approved and marketed in South Korea for OA since 2003. We update the FDA annually on safety data generated by Apimeds Korea from South Korea.
We aim to obtain FDA approval for Apitox in the United States market for treatment of inflammation and pain management symptoms associated with knee OA, and eventually MS, and expand the indication portfolio in the autoimmune market with a strategic marketing partner. The marketing partner strategy is common in the pharmaceutical marketplace, as the infrastructure, overhead, and barriers to entry dilute the focus and can rapidly erode the financial well-being of small, product development-based companies such as us. By identifying the strategic marketing partner at an early stage, the companies can deliver a final product, or family of products, in a form factor or variety of form factors over time, that specifically suit the target market. We believe that Apitox represents a significant opportunity as a platform technology, with numerous product-line extensions, and the potential for new, ancillary products such as delivery devices.
Reimbursement Strategy
Apimeds expects to apply to the Centers for Medicare and Medicaid Studies (“CMS”) for temporary generic reimbursement codes 12 to 18 months prior to a BLA approval. Temporary codes are used until manufacturers apply for, and receive, permanent codes, which identify the drug and its therapeutic class. Permanent codes are issued by CMS on a rolling quarterly basis.
11
We will engage third party contractors to assist the us with reimbursement, coding and policy development prior to, during and at the time of approval of Apitox. We will look for a contractor to provide the following services to us:
| ● | Coding Assessment and Strategy/Execution — CPT Review of Apitox Administration by Multiple Intradermal Injections. Assess the landscape to ensure a clear understanding of the key dynamics and analyze relevant proxies and precedent. Further assess relevant drug administration codes and whether appropriate codes exist. |
| ● | Medical Coverage Policy Analysis — Provide a framework and set expectations for Medicare’s anticipated coverage approach to Apitox, specifically in the context of intra articular hyaluronic acid use agent coverage policies and implications of their efficacy uncertainty. |
| ● | Medicare Local Coverage Analysis and Implications — Given the significance of Medicare policy standards, local and national Medicare policies often shape payer and provider perceptions and decisions. As complex statutory and regulatory guidance shape Medicare decision-making, ADVI analyzes, investigates, and synthesize Medicare policies that could affect access (coverage, coding and reimbursement) for Apitox. |
| ● | Medicaid and Commercial Coverage Analysis and Implications — Analyze available medical policies for five large state Medicaid agencies (based on population and geographic variation) and major commercial payers (where publicly available). |
| ● | Payer Policy Internal Expert Interviews — Conduct payer interviews with relevant Medicare, Medicaid and commercial policy advisors. |
| ● | HCPCS Coding and Payment Assessment — Assess the coding and reimbursement landscape to ensure Apimeds has a clear understanding of the key dynamics with the HCPCS application process and the Medicare Hospital Outpatient Prospective Payment System (OPPS) pass-through status application process. Through this assessment, identify the areas of concern, expectations, timing, timelines, and processes associated. This is especially relevant given the 2020 implementation of a new HCPCS review process. |
| ● | Address key Part B/medical benefit implications to Apitox in the following fields: |
| ● | HCPCS and OPPS application timelines (and potential evolution leading to launch), |
| ● | coding/access implications prior to code assignment (e.g., NOC/miscellaneous codes), review the merits/risks of Q-code, |
| ● | further review the application processes, expectations, case examples, timelines, and hurdles that APUS may face across settings of care, payers, and with CMS, |
| ● | case examples, timelines, and hurdles across settings of care with payers and CMS, |
| ● | review of reimbursement implications, and |
| ● | methodologies (ASP, WAC, AWP), role of sequestration, 340B, patient financial burden. |
| ● | Develop Payer (with Emphasis on Medicare) Launch Recommendations — Based on the above primary and secondary research, synthesize the discussions and summarize the overall findings of the payer survey, highlighting themes, and provide recommendations and considerations for optimizing market access, given the current and evolving reimbursement landscape. This section will include payer (emphasis on Medicare) launch strategy recommendations (including timeline) and a local/national Medicare engagement strategy. |
Competition
We compete in an industry characterized by rapidly advancing technologies, intense competition, a changing regulatory and legislative landscape and a strong emphasis on the benefits of intellectual property protection and regulatory exclusivities.
12
Like any biopharmaceutical company, we face competition from multiple sources, including large or established pharmaceutical, biotechnology, and wellness companies, academic research institutions, government agencies, and private institutions. We believe our drug candidate will prevail amid the competitive landscape through its efficacy, safety, administration methods, cost, public and institutional demand, intellectual property portfolio, and treatment of the root cause of many age-associated diseases.
Many of our competitors, either alone or with strategic partners, have substantially greater financial, technical, and human resources than we do. Accordingly, our competitors may be more successful in obtaining approval for treatments and achieving widespread market acceptance, rendering our treatments obsolete or non-competitive. Accelerated merger and acquisition activity in the biotechnology and biopharmaceutical industries may result in even more resources concentrated among a smaller number of our competitors. These companies also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical study sites, patient registration for clinical studies, and acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunity could be substantially limited in the event that our competitors develop and commercialize products that are more effective, safer, more tolerable, more convenient, or less expensive than our comparable products. In geographies that are critical to our commercial success, competitors may also obtain regulatory approvals before us, resulting in our competitors building a strong market position in advance of our products’ entry. We believe the factors determining the success of our programs will be the efficacy, safety, and convenience of our drug candidates.
Additionally, consumer preference for branded, generic or private label products sold by competitors could adversely impact our financial performance. Our competitors, which differ within individual geographic markets, include large-scale retailers, smaller high-growth companies (which often operate on a regional basis and offer aggressive competition), multinational corporations moving into or expanding their presence in the consumer healthcare market, and “private-label” products sold by retailers.
Our aim is to reduce the use of NSAIDS and opioid use as it relates to the pain management associated with OA. We believe that if approved by the FDA, Apitox may be a non-addictive option to patients experiencing debilitating pain.
Business Agreement
On August 2, 2021, we entered into an agreement with Apimeds Korea, a principal stockholder of the Company (the “Business Agreement”). Pursuant to the Business Agreement, Apimeds Korea granted to the Company a sublicensable, royalty-bearing license to utilize all prior clinical development data associated with Apitoxin, Apitox, and all related names, advance clinical research, develop, manufacture and commercialize and sell Apitox in the United States. In exchange for this license, the Company will pay Apimeds Korea a perpetual royalty of 5% of the Company’s earnings before interest and taxes (as determined consistent with GAAP, derived from the sale or license of Apitox, less any shipping, handling, and insurance charges, credits (arising from returns or other adjustments), discounts, rebates, or allowances of any kind (if any). The Business Agreement can be terminated by mutual written agreement by the parties and will automatically terminate upon the bankruptcy or dissolution of the Company.
Assignment Agreement
On October 12, 2021, we entered into an intellectual property assignment agreement (the “Assignment Agreement”), which was effective as of May 12, 2020, with Apimeds Korea and Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and the founder of Apimeds Korea. During Dr. Kim’s engagement with Apimeds Korea, he contributed to the development of the intellectual property as it relates to Apitoxin, which will be marketed in the United States as Apitox (the “Assigned IP”).
Pursuant to the Assignment Agreement, Dr. Kim sold, transferred, and conveyed all his rights, title and interest in the Assigned IP to Apimeds Korea. Dr. Kim retained no right to use the Assigned IP. Additionally, the Assignment Agreement acknowledged that the Assigned IP was licensed to us to use via the Business Agreement.
Intellectual Property
Apitox’s API is bee venom, a natural, non-synthetic compound that is not patentable, so we rely principally on trade secrets to protect our rights to Apitox, particularly the method and process of manufacturing Apitox.
13
Supplier
We purchase venom from our United States supplier, Apico, Inc. (“Apico”), via a letter agreement. Pursuant to the letter agreement, Apico agreed that for a period of ten years, or until November 3, 2031 it would not supply Apis Mellifea venom for pharmaceutical use for any buyer other than us; provided that Apico may also supply Apimeds Korea for its use outside of the United States. The letter agreement excludes customers using venom for immunology, cosmetic or any other “non-pharmaceutical” use. The letter agreement may be terminated upon mutual written consent of both Apico and the Company.
Apico has developed and practices a proprietary method of harvesting venom. It operates under and is certified in current good manufacturing practice regulations enforced by the FDA and has an active and current Drug Master File (“DMF”) with the FDA. DMF’s are submissions to the FDA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of human drug products. We have an exclusive relationship with our supplier for pharmaceutical use in the United States and they are not permitted to sell to any other party for pharmaceutical use.
Apimeds Korea has a number of proprietary analytical methods for the classification and identification of specific pharmacologically active fractions of its venom, along with numerous manufacturing processes from filtration, vial filing and lyophilization required to produce Apitoxin. Apitoxin is the only approved and commercially available therapeutic product containing purified and sterile bee venom that is registered as an API in South Korea. The proprietary methods developed and practiced for the commercial manufacturing of Apitoxin include dilution, filtering, vial staging and lyophilization parameters and cycles.
We plan to file Apitox as a BLA with the Centers for Biologics and Research of the FDA following the successful completion of our Phase III trial for knee OA. The FDA provides 12-year market exclusivity at the time of approval of a BLA, with the potential for a six-month extension upon approval for pediatric use. If the BLA is approved, the 12-year period would be retroactive to the date of the application.
We intend to file a U.S. trademark application for “Apitox”.
Regulatory Environment
Government Regulation and Product Approval
In the United States, biological products are subject to regulation under the Federal Food, Drug, and Cosmetic Act (the “FDCA”), and the Public Health Service Act (the “PHSA”), and other federal, state, and local statutes and regulations. Both the FDCA and PHSA and their corresponding regulations govern, among other things, the research, development, clinical trials, testing, manufacturing, quality control, safety, purity and potency (efficacy), labeling, packaging, storage, record keeping, distribution, reporting, marketing, promotion, advertising, post-approval monitoring, and post-approval reporting involving biological products. Along with third-party contractors, we will be required to navigate the various preclinical and clinical regulatory obligations and the commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidate. The processes for obtaining regulatory approvals in the United States, along with subsequent compliance with applicable laws and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Government policies may change, and additional government regulations may be enacted that could prevent or delay further development or regulatory approval of any product candidates, product or manufacturing changes, additional disease indications or label changes. We cannot predict the likelihood, nature or extent of government regulation that might arise from future legislative or administrative action.
Review and Approval for Licensing Biologics in the United States
In the United States, FDA regulates our current product candidate as a biological product, or biologics, under the FDCA, the PHSA, and associated implementing regulations. Biologics, like other drugs, are used for the diagnosis, cure, mitigation, treatment, or prevention of disease in humans. In contrast to low molecular weight drugs, which have a well-defined structure and can be thoroughly characterized, biologics are generally derived from living material (human, animal, or microorganism), are complex in structure, and thus are usually not fully characterized.
14
Biologics are also subject to other federal, state, and local statutes and regulations. The failure to comply with applicable statutory and regulatory requirements at any time during the product development process, approval process, or after approval may subject a sponsor or applicant to administrative or judicial enforcement actions. These actions could include the suspension or termination of clinical trials by FDA, FDA’s refusal to approve pending applications or supplemental applications, withdrawal of an approval, issuance of warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, import detention, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by FDA, the Department of Justice (“DOJ”), and other governmental entities.
An applicant seeking approval to market and distribute a biologic in the United States must typically undertake the following:
| ● | completion of non-clinical laboratory tests and studies performed in accordance with FDA’s good laboratory practice (“GLP”) regulations; |
| ● | manufacture, labeling and distribution of investigational drugs in compliance with FDA’s current good manufacturing practice (“cGMP”) requirements; |
| ● | submission to FDA of an investigational new drug application (“IND”), which must become effective before clinical trials may begin and must be updated annually and when significant changes are made; |
| ● | approval by an independent institutional review board (“IRB”) for each clinical site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with FDA’s Good Clinical Practices (“GCP”) to establish the safety, purity, and potency of the proposed biological product candidate for its intended purpose; |
| ● | after completion of all pivotal clinical trials, preparation of and submission to FDA of a BLA requesting marketing approval, which includes providing sufficient evidence to establish the efficacy, safety, purity, and potency of the proposed biological product for its intended use, including from results of nonclinical testing and clinical trials; |
| ● | satisfactory completion of an FDA advisory committee review, when appropriate, as may be requested by FDA to assist with its review; |
| ● | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the proposed product, or certain components thereof, are produced to assess compliance with cGMP and data integrity requirements to assure that the facilities, methods, and controls are adequate to preserve the biological product’s identity, strength, quality, and purity and, if applicable, FDA’s good tissue practice (“GTP”) requirements for human cellular and tissue products; |
| ● | satisfactory completion of FDA inspections of selected clinical investigation sites to assure compliance with GCP requirements and the integrity of the clinical data; |
| ● | satisfactory completion of an FDA sponsor GCP inspection, often conducted at the applicant’s headquarters facility; |
| ● | payment of user fees (unless there is a waiver, exemption, or reduction) under the Prescription Drug User Fee Act (“PDUFA”) for the relevant year; |
| ● | FDA’s review and approval of the BLA to permit commercial marketing of the licensed biologic for particular indications for use in the United States; |
| ● | compliance with post-approval requirements, including the potential requirements to implement a risk evaluation and mitigation strategy (“REMS”), to report adverse events and biological product deviations, and to complete any post-approval studies; and |
| ● | completion of any post-approval clinical studies required by FDA, such as confirmatory trials or pediatric studies. |
15
From time to time, legislation is drafted, introduced, and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing, and marketing of biological products regulated by FDA. In addition to new legislation, FDA regulations, guidance documents, and policies are often revised or interpreted by the agency in ways that may significantly affect the regulation of biological products in the United States. It is impossible to predict whether further legislative changes will be enacted or whether FDA regulations, guidance, policies, or interpretations will change, and the effects of any such changes.
Preclinical and Clinical Development
Before an applicant can begin testing the potential product candidate in human subjects, the applicant must first conduct preclinical studies. Preclinical studies may include laboratory evaluations of product chemistry, toxicity, and formulation, as well as in vitro and animal studies to assess the potential safety and activity of the drug for initial testing in humans and to establish a rationale for therapeutic use. Preclinical studies are subject to federal regulations and requirements, including GLP regulations, which govern the conduct of animal studies designed to test a product’s safety. None of our preclinical studies to date have been animal studies. The results of an applicant’s preclinical studies are submitted to FDA as part of an IND.
An IND is a request for authorization from FDA to administer an investigational new drug product to humans. An IND is an exemption from the FDCA that allows an unapproved drug to be shipped in interstate commerce for use in a clinical trial. Such authorization must be secured prior to interstate shipment and administration of a biological drug that is not subject of an approved BLA. In support of an IND, applicants must submit a protocol for each clinical trial, which details, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments.
Human clinical trials may not begin until an IND is effective. The IND automatically becomes effective 30 days after receipt by FDA, unless FDA raises safety concerns or questions about the proposed clinical trial within the 30-day time period. In such a case, FDA may place the IND on clinical hold and the IND sponsor must resolve any of FDA’s outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in regulatory authorization to begin a clinical trial.
FDA may also place a clinical hold or partial clinical hold on a clinical trial following commencement of the trial under an IND. A clinical hold is an order issued by FDA to the sponsor to delay a proposed clinical investigation or to suspend an ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical work requested under the IND. For example, under a partial clinical hold, FDA may instruct a sponsor not to enroll any new patients into a study but permit the previously enrolled patients to continue in the study. No more than 30 days after imposition of a clinical hold or partial clinical hold, FDA will provide the sponsor a written explanation of the basis for the hold. Following issuance of a clinical hold or partial clinical hold, an investigation may only resume after the FDA has notified the sponsor that the investigation may proceed. FDA will base that determination on information provided by the sponsor addressing the deficiencies previously cited or otherwise satisfying FDA that the investigation can proceed.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP regulations, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. If a sponsor chooses to conduct a foreign clinical study under an IND, all FDA IND requirements must be met unless waived. When the foreign clinical study is not conducted under an IND, the sponsor must ensure that the study complies with GCP regulations in order to use the study as support for an IND or application for marketing approval, including review and approval by an IRB and informed consent from subjects.
Furthermore, an independent IRB for all sites participating in a clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at each site and must monitor the trial until completed. Regulatory authorities, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives.
16
Some trials also include oversight by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board (“DSMB”). DSMBs review unblinded study data at pre-specified times during the course of the study. If the DSMB determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy, the DSMB can make a recommendation to the sponsor to modify or stop the trial.
Other grounds for a sponsor’s decision to suspend or terminate a study may be made based on evolving business objectives or the competitive climate.
For purposes of BLA approval, clinical trials are typically conducted in the following sequential phases:
| ● | Phase 1: The investigational product is initially introduced into a small group of healthy human subjects or patients with the target disease or condition. These trials are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans and the side effects associated with increasing doses. These trials may also yield early evidence of effectiveness. |
| ● | Phase 2: The investigational product is administered to a slightly larger patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages, and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase III clinical trials. |
| ● | Phase 3: The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to generate sufficient data to statistically demonstrate the efficacy and safety of the product, to establish the overall risk/benefit ratio of the investigational product, and to provide an adequate basis for product approval by FDA. |
These phases may overlap or be combined. In some cases, FDA may require, or companies may voluntarily pursue, additional clinical trials after a product are approved to gain more information about the product, referred to as Phase 4 trials. Post-approval trials are conducted following initial approval, often to develop additional data and information relating to the use of the product in new indications.
Progress reports detailing the results of the clinical trials must be submitted at least annually to FDA. In addition, IND safety reports must be submitted to FDA for any of the following: serious and unexpected suspected adverse reactions in study subjects; findings from epidemiological studies, pooled analysis of multiple studies, animal or in vitro testing, or other clinical studies, whether or not conducted under an IND, and whether or not conducted by the sponsor, that suggest a significant risk in humans exposed to the drug; and any clinically important increase in the rate of a serious suspected adverse reaction over such rate listed in the protocol or investigator brochure.
A sponsor’s planned clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. FDA will typically inspect one or more clinical sites to assure compliance with GCP and the integrity of the clinical data submitted.
During clinical development, the sponsor often refines the indication and endpoints on which the BLA will be based. For endpoints based on patient-reported outcomes (“PROs”), the process typically is an iterative one. FDA has issued guidance on the framework it uses to evaluate PRO instruments. Although the agency may offer advice on optimizing PRO instruments during the clinical development process, FDA usually reserves final judgment until it reviews the BLA.
Concurrent with clinical trials, companies often complete additional animal studies, and develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, must develop methods for testing the identity, strength, quality, purity and potency of the final drug. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
17
BLA Submission and Review
Assuming successful completion of all required clinical testing in accordance with all applicable regulatory requirements, an applicant may submit a BLA requesting licensing to market the biologic for one or more indications in the United States. The BLA must include the results of nonclinical studies and clinical trials; detailed information on the product’s chemistry, manufacture, controls; and proposed labeling. Under the PDUFA, a BLA submission is subject to an application user fee, unless a waiver, reduction, or exemption applies.
FDA will initially review the BLA for completeness before accepting it for filing. Under FDA’s procedures, the agency has 60 days from its receipt of a BLA to determine whether the application will be accepted for filing and substantive review. If the agency determines that the application does not meet this initial threshold standard, FDA may refuse to file the application and request additional information, in which case the application must be resubmitted with the requested information and review of the application delayed.
After the BLA is accepted for filing, FDA reviews the BLA to determine, among other things, whether a product is safe, pure, and potent and if the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued identity, strength, quality, safety, purity, and potency. To ensure cGMP, GLP, GCP, GTP, and other regulatory compliance, an applicant must incur significant expenditure of time, money, and effort in the areas of training, record keeping, production and quality control. In addition, FDA expects that all data be reliable and accurate, and requires sponsors to implement meaningful and effective strategies to manage data integrity risks. Data integrity is an important component of the sponsor’s responsibility to ensure the safety, efficacy and quality of its product or products.
For cellular products, FDA will not approve the product if the manufacturer is not in compliance with the GTPs, to the extent applicable. GTPs are FDA regulations and guidance documents that govern the methods used in, and the facilities and controls used for, the manufacture of human cells, tissue, and cellular and tissue-based products (“HCT/Ps”), which are human cells or tissue intended for implantation, transplant, infusion, or transfer into a human recipient. The primary intent of the GTP requirements is to ensure that cell and tissue-based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. FDA regulations also specify how HCT/P establishments must register and list their HCT/Ps with FDA and how they must evaluate donors through screening and testing, where applicable.
If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The performance goals and policies implemented by FDA under the PDUFA generally provide for FDA action on an original BLA within 10 months of filing, which (as discussed above) typically occurs within 60 days of submission, but that deadline is extended in certain circumstances. Furthermore, the review process is often significantly extended by FDA’s requests for additional information or clarification.
FDA may refer applications for novel products or products that present difficult questions of safety or efficacy to an advisory committee. Typically, an advisory committee consists of a panel that includes clinicians and other experts who will review, evaluate, and provide a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions and usually has followed such recommendations.
After FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its components will be produced, FDA may issue an approval letter or a Complete Response Letter (“CRL”). An approval letter authorizes commercial marketing of the biological with specific prescribing information for specific indications. A CRL will describe all of the deficiencies that FDA has identified in the BLA, except that where FDA determines that the data supporting the application are inadequate to support approval, FDA may issue the CRL without first conducting required inspections, testing submitted product lots and/or reviewing proposed labeling. If and when the deficiencies have been addressed to FDA’s satisfaction in a resubmission of the BLA, FDA will issue an approval letter. In issuing the CRL, the FDA may recommend actions that the applicant might take to place the BLA in condition for approval, including requests for additional data, information, or clarification. FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied and may require additional testing or information and/or require new clinical trials. Even with submission of this additional information, FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
18
During the approval process, FDA will determine whether a REMS is necessary to help ensure the benefits outweigh the risks of the biologic. A REMS is a safety strategy to manage a known or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. If FDA concludes that a REMS is needed, the BLA sponsor must submit a proposed REMS and FDA will not approve the BLA without a REMS that the agency has determined is acceptable.
If the FDA approves a product, it may limit the approved indications for use for the product, or require that contraindications, warnings, or precautions be included in the product labeling. FDA may also require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess the drug’s safety after approval. FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs.
FDA may also require testing and surveillance programs to monitor the product after commercialization. For biologics, such testing may include official lot release, which requires the manufacturer to perform certain tests on each lot of the product before it is released for distribution. The manufacturer then typically must submit samples of each lot of products to the FDA, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products itself, before releasing the lots for distribution by the manufacturer.
In general, an approved BLA only allows the sponsor to market the biologic as approved, without modification. If, for example, a sponsor modifies an approved T cell product to target different peptides or in our case to target another HLA type, the sponsor would be required to either file a supplemental BLA with FDA or receive FDA approval for a comparability protocol in order to implement this change into the final product.
The FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace.
Post-Approval Requirements
Any products manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, reporting of certain deviations and adverse experiences, product sampling and distribution, and advertising and promotion of the product. After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are often subject to further testing requirements and FDA review and approval, depending on the nature of the post-approval change. There also are continuing user fee requirements, under which FDA assesses an annual program fee for each product identified in an approved BLA. Biologic manufacturers and their third-party contractors are required to register their facilities with the FDA and certain state agencies. These facilities are subject to routine and periodic unannounced inspections by FDA and certain state agencies for compliance with cGMP, post-marketing safety reporting and data integrity requirements, which impose certain procedural and documentation requirements to assure quality of manufacturing and product. FDA has increasingly observed cGMP violations involving data integrity during site inspections and is a significant focus of its oversight. Requirements with respect to data integrity include, among other things, controls ensuring complete and secure data; activities documented at the time of performance; audit trail functionality; authorized access and limitations; validated computer systems; and review of records for accuracy, completeness, and compliance with established standards.
Post-approval changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon the sponsor and any third-party manufacturers that the sponsor may use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain compliance with cGMP, data integrity, pharmacovigilance, and other aspects of regulatory compliance.
19
The FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-approval studies to assess new safety risks; or imposition of distribution or other restrictions under a REMS. Other potential consequences include, for example:
| ● | restrictions on the marketing or manufacturing of a product, complete withdrawal of the product from the market, or product recalls; |
| ● | fines, warning or untitled letters, or holds on post-approval clinical studies; |
| ● | refusal of FDA to approve pending applications or supplements to approved applications, or suspension or revocation of existing product approvals; |
| ● | product seizure or detention, or refusal of FDA to permit the import or export of products; or |
| ● | permanent injunctions and consent decrees, including the imposition of civil or criminal penalties. |
FDA strictly regulates the marketing, labeling, advertising, and promotion of prescription drug products placed on the market. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved labeling. FDA’s regulation includes, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities and promotional activities involving the Internet and social media. Promotional claims relating to a product’s safety or effectiveness are prohibited before the drug is approved. After approval, a product generally may not be promoted for uses that are not approved by FDA, as reflected in the product’s prescribing information. In the United States, healthcare professionals are generally permitted to prescribe drugs for such uses not described in the drug’s labeling, known as off-label uses, because FDA does not regulate the practice of medicine. However, FDA regulations impose rigorous restrictions on manufacturers’ communications and prohibit the promotion of off-label uses. It may be permissible, under very specific, narrow conditions, for a manufacturer to engage in non-promotional, non-misleading communication regarding off-label information, such as distributing scientific or medical journal information.
If a company is found to have promoted off-label uses, it may become subject to adverse public relations and administrative and judicial enforcement by FDA, the DOJ, or the Office of the Inspector General of the Department of Health and Human Services (“HHS”), as well as other federal and state authorities. This could subject a company to a range of penalties that could have a significant commercial impact, including civil, administrative, and criminal fines, penalties, and agreements that materially restrict the manner in which a company promotes or distributes products. The federal government has levied large civil, administrative, and criminal fines and penalties against companies for alleged improper promotion and has also requested that companies enter into Corporate Integrity Agreements and Consent Decrees of Permanent Injunction under which specified promotional conduct is changed or curtailed.
The distribution of prescription drugs and biologics are subject to the Drug Supply Chain Security Act (“DSCSA”), which requires manufacturers and other stakeholders to comply with product identification, tracing, verification, detection and response, notification, and licensing requirements. In addition, the Prescription Drug Marketing Act and its implementing regulations and state laws limit the distribution of prescription pharmaceutical product samples, and the DSCSA imposes requirements to ensure accountability in distribution and to identify and remove prescription drug and biological products that may be counterfeit, stolen, contaminated, or otherwise harmful from the market.
Expedited Development and Review Programs
FDA offers a number of expedited development and review programs for qualifying product candidates. The fast-track program is intended to expedite or facilitate the process of reviewing new products that meet certain criteria. Specifically, new products are eligible for fast-track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. A product intended to treat a serious or life-threatening disease or condition may also be eligible for breakthrough therapy designation to expedite its development and review. Any marketing application for a biologic submitted to FDA for approval, including a product with a fast-track designation and/or breakthrough therapy designation, may be eligible for other types of FDA programs intended to expedite FDA review and approval process, such as priority review and accelerated approval. FDA also may grant accelerated approval to certain products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions.
20
The RMAT designation, which we are currently planning to seek for some of our therapies, is intended to facilitate an efficient development program for, and expedite review of, any drug that meets the following criteria: (1) the drug is a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with limited exceptions; (2) the drug is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (3) preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such a disease or condition. Like breakthrough therapy designation, RMAT designation provides potential benefits that include more frequent meetings with FDA to discuss the development plan for the product candidate and eligibility for rolling review and priority review. Products granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites (including through expansion to additional sites) so as to remove any likelihood of site-specific or investigator-specific bias on the evidence of effectiveness. Once approved, when appropriate, FDA can permit fulfillment of post-approval requirements for RMATs receiving accelerated approval through the submission of clinical evidence, clinical studies, patient registries, or other sources of real-world evidence such as electronic health records; through the collection of larger confirmatory datasets; or through post-approval monitoring of all patients treated with the therapy prior to approval.
Fast track designation, breakthrough therapy designation, priority review, accelerated approval, and RMAT designation do not change the standards for approval but may expedite the development or approval process.
Patent Term Restoration and Marketing Exclusivity
After approval, owners of relevant drug or biological product patents may apply for up to a five year term patent extension to restore a portion of patent term lost during product development and FDA review of a BLA if approval of the application is the first permitted commercial marketing or use of a drug or biologic containing the active ingredient under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The allowable patent term extension is calculated as one-half of the product’s testing phase, which is the time between the effective date of an IND and initial BLA submission, and all of the approval phase, which is the time between BLA submission and approval, up to a maximum of five years. The time can be shortened if the FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years from the date of FDA approval of the product. Only one patent claiming each approved product is eligible for restoration and the patent holder must apply for restoration within 60 days of approval, even if the product cannot be commercially marketed at that time. The USPTO, in consultation with FDA, reviews and approves the application for patent term restoration.
For patents that might expire during the BLA application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the USPTO must determine that approval of the product candidate covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a product candidate for which a BLA has not been submitted.
Biosimilars and Marketing Exclusivities
The Biologics Price Competition and Innovation Act (“BPCIA”) created an abbreviated approval pathway for biological product candidates shown to be highly similar to or interchangeable with an FDA licensed biological product. A biological product on which another biological product candidate’s BLA relies to establish bio similarity is known as a reference product. Bio similarity sufficient to reference a prior FDA-approved product requires that there be no differences in conditions of use, route of administration, dosage form and strength, and no clinically meaningful differences between the biological product candidate and the reference product in terms of safety, purity, and potency. Bio similarity must be shown through analytical trials, animal trials and at least one clinical trial, unless the Secretary of HHS waives a required element. A biosimilar product candidate may be deemed interchangeable with a prior approved product if it meets the higher hurdle of demonstrating that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biological product candidate and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. Complexities associated with the larger, and often more complex, structures of biologics, as well as the process by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being resolved by FDA.
21
A reference biologic is granted 12 years of exclusivity from the time of first licensure of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biological product candidate submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against a finding of interchangeability for other biologics for the same condition of use for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) 18 months after the first interchangeable biosimilar is approved if there is no patent challenge, (iii) 18 months after resolution of a lawsuit over the patents of the reference biologic in favor of the first interchangeable biosimilar applicant, or (iv) 42 months after the first interchangeable biosimilar’s application has been approved if a patent lawsuit is ongoing within the 42 month period. At this time, it is unclear whether products deemed “interchangeable” by FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy laws and regulations.
Healthcare Regulation
Coverage, Pricing, and Reimbursement
Our ability to successfully commercialize any products for which we receive regulatory approval for commercial sale will depend, in part, on the extent to which third-party payors provide coverage and establish adequate reimbursement levels for such products, and significant uncertainty exists as to the coverage and reimbursement status of any products for which may we obtain regulatory approval. In the United States, third-party payors include federal and state health care programs, private managed care providers, health insurers and other organizations. The process for determining whether a third-party payor will provide coverage for a product may be separate from the process for setting the price of a product or for establishing the reimbursement rate that such a payor will pay for the product. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication. Third-party payors are increasingly challenging the price, examining the medical necessity, and reviewing the cost-effectiveness of medical products, therapies, and services, in addition to questioning their safety and efficacy. We may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our product candidates may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
The marketability of any product candidates for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on healthcare pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Compliance Requirements
Although we currently do not have any commercialized products, our current and future business operations may be subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security, price reporting and physician sunshine laws. Some of our pre-commercial activities are subject to some of these laws.
22
The federal Anti-Kickback Statute makes it illegal for any person or entity, including a prescription drug manufacturer or a party acting on its behalf to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration in cash or in kind that is intended to induce or reward the referral of business, including the purchase, order, or lease of any item or service for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, formulary managers and beneficiaries on the other.
Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have found that the Anti-Kickback Statute may be violated if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare program business. In addition, liability may be established without actual knowledge of the statute or specific intent to violate it. Violations of this law are punishable by up to ten years in prison, and can also result in criminal fines, civil money penalties and exclusion from participation in federal healthcare programs.
Moreover, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
The federal civil False Claims Act prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly making, using, or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. Persons and entities can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. Many pharmaceutical and other healthcare companies have been investigated and have reached substantial financial settlements with the federal government under the civil False Claims Act for a variety of alleged improper marketing activities, including: providing free product to customers with the expectation that the customers would bill federal programs for the product; providing sham consulting fees, grants, free travel and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which are used to set drug payment rates under government healthcare programs. Penalties for federal civil False Claims Act violations may include up to three times the actual damages sustained by the government, plus mandatory civil penalties of between $13,508 and $27,018 for each separate false claim, and the potential for exclusion from participation in federal healthcare programs. In addition, although the federal False Claims Act is a civil statute, False Claims Act violations may also implicate various federal criminal statutes.
The healthcare fraud provisions of the Health Insurance Portability and Accountability Act (“HIPAA”) prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Many states have analogous laws and regulations, such as: state anti-kickback and false claims laws that may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to certain healthcare providers; laws that require drug manufacturers to report information related to clinical trials or information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; laws that restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs; and laws and local ordinances that require identification or licensing of sales representatives.
23
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their implementing regulations, mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. Among other things, HITECH makes HIPAA’s security standards directly applicable to business associates, defined as independent contractors or agents of covered entities that create, receive, or obtain protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities and business associates and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties.
The U.S. federal Physician Payment Sunshine Act, implemented as the Open Payments Program, requires manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to direct or indirect payments and other transfers of value to physicians and teaching hospitals (and certain other practitioners as of 2022), as well as ownership and investment interests held in the company by physicians and their immediate family members.
Because we intend to commercialize products that could be reimbursed under a federal health care program and other governmental healthcare programs, we intend to develop a comprehensive compliance program that establishes internal control to facilitate adherence to the rules and program requirements to which we will or may become subject. Although the development and implementation of compliance programs designed to establish internal control and facilitate compliance can mitigate the risk of investigation, prosecution, and penalties assessed for violations of these laws, the risks cannot be entirely eliminated.
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and individual imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Health Care Reforms
In the United States and some foreign jurisdictions, there have been, and continue to be, legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities, and affect the ability to profitably sell product candidates for which marketing approval is obtained. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
For example, the Affordable Care Act (“ACA”) substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA contains provisions that may reduce the profitability of drug products through increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. The ACA made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate. The ACA also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and by enlarging the population potentially eligible for Medicaid drug benefits.
24
There have been judicial challenges to certain aspects of the ACA, as well as efforts by Congress to modify, and by agencies to alter the implementation of, certain aspects of the ACA. For example, Congress eliminated the tax penalty for failure to comply with the ACA’s individual mandate to carry health insurance. Further, the Bipartisan Budget Act of 2018, among other things, amended the ACA to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D to close the coverage gap in most Medicare drug plans, commonly referred to as the donut hole.
It is possible that the ACA, as currently enacted or as may be amended in the future, as well as other healthcare reform measures, including those that may be adopted in the future, may result in more rigorous coverage criteria, and less favorable payment methodologies, or other downward pressure on coverage and payment and the price that we receive for any approved product. Any reduction in reimbursement or restriction on coverage under Medicare or other federal health care programs may result in a similar reduction or restriction by private payors.
Other legislative changes have been proposed and adopted in the U.S. since the ACA was enacted. For example, the Inflation Reduction Act introduces several changes to the Medicare Part D benefit, including a limit on annual out-of-pocket costs and a change in manufacturer liability under the program which could negatively affect the profitability of our product candidates. The IRA sunsets the current Part D coverage gap discount program starting in 2025 and replaces it with a new manufacturer discount program. Failure to pay a discount under this new program will be subject to a civil monetary penalty. In addition, the IRA establishes a Medicare Part B inflation rebate scheme effective January 2023 and a Medicare Part D inflation rebate scheme effective October 2022, under which, generally speaking, manufacturers will owe rebates if the price of a Part B or Part D drug increases faster than the pace of inflation. Failure to timely pay a Part B or D inflation rebate is subject to a civil monetary penalty. The IRA also creates a drug price negotiation program under which the prices for Medicare units of certain high Medicare spend drugs and biologicals without generic or biosimilar competition will be capped by reference to, among other things, a specified non-federal average manufacturer price starting in 2026. Failure to comply with requirements under the drug price negotiation program is subject to an excise tax and/or a civil monetary penalty. Congress continues to examine various policy proposals that may result in pressure on the prices of prescription drugs with respect to the government health benefit programs and otherwise. The IRA or other legislative changes could impact the market conditions for our product candidates.
In general, there has been heightened governmental scrutiny over the manner in which drug manufacturers set prices for their commercial products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Drug Pedigree Laws
State and federal governments have proposed or enacted various drug pedigree laws which can require the tracking of all transactions involving prescription drugs from the manufacturer to the pharmacy (or other dispensing) level. Companies are required to maintain records documenting the chain of custody of prescription drug products beginning with the purchase of such products from the manufacturer. Compliance with these pedigree laws requires implementation of extensive tracking systems as well as heightened documentation and coordination with customers and manufacturers. While we fully intend to comply with these laws, there is uncertainty about future changes in legislation and government enforcement of these laws. Failure to comply could result in fines or penalties, as well as loss of business that could have a material adverse effect on our financial results.
Federal Regulation of Patent Litigation Settlements and Authorized Generic Arrangements
As part of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, companies are required to file with the U.S. Federal Trade Commission (“FTC”) and the U.S. Department of Justice certain types of agreements entered into between brand and generic pharmaceutical companies related to the settlement of patent litigation or manufacture, marketing and sale of generic versions of branded drugs. This requirement could affect the manner in which generic drug manufacturers resolve intellectual property litigation and other disputes with brand pharmaceutical companies and could result generally in an increase in private-party litigation against pharmaceutical companies or additional investigations or proceedings by the FTC or other governmental authorities.
25
Other
The U.S. federal government, various states and localities have laws regulating the manufacture and distribution of pharmaceuticals, as well as regulations dealing with the substitution of generic drugs for branded drugs. Our operations are also subject to regulation, licensing requirements and inspection by the states and localities in which our operations are located or in which we conduct business.
Certain of our activities are also subject to FTC enforcement actions. The FTC enforces a variety of antitrust and consumer protection laws designed to ensure that the nation’s markets function competitively, are vigorous, efficient and free of undue restrictions. Federal, state, local and foreign laws of general applicability, such as laws regulating working conditions, also govern us.
In addition, we are subject to numerous and increasingly stringent federal, state and local environmental laws and regulations concerning, among other things, the generation, handling, storage, transportation, treatment and disposal of toxic and hazardous substances, the discharge of pollutants into the air and water and the cleanup of contamination. We are required to maintain and comply with environmental permits and controls for some of our operations, and these permits are subject to modification, renewal and revocation by the issuing authorities. Our environmental capital expenditures and costs for environmental compliance may increase in the future as a result of changes in environmental laws and regulations or increased manufacturing activities at any of our facilities. We could incur significant costs or liabilities as a result of any failure to comply with environmental laws, including fines, penalties, third-party claims and the costs of undertaking a clean-up at a current or former site or at a site to which our wastes were transported. In addition, we have grown in part by acquisition, and our diligence may not have identified environmental impacts from historical operations at sites we have acquired in the past or may acquire in the future.
Employees
As of the date of this Annual Report, we have seven (7) full time employees. We have no part-time employees and we engage one consultant. We believe that we maintain good relations with our employees.
INFORMATION ABOUT THE BUSINESS OF MINDWAVE INNOVATIONS INC
Unless the context otherwise indicates or requires, all references in this section to “we,” “us,” “our,” “our company” “the Company” and “MindWave” refer to MindWave Innovations Inc
MindWave Innovations Inc (“MindWave,” the “Company,” “we,” “us” or “our”) is a technology platform company focused on institutional Digital Asset Treasury (“DAT”) solutions, centered on enabling corporations and institutions to hold, manage, and generate yield on Bitcoin reserves through a compliant, scalable infrastructure. Our strategic model integrates secure digital treasury wallets, AI-supported Bitcoin yield programs, and a validator-enabled ecosystem supported by our native token, $NILA. TechyTrade Innovations Pte. Ltd. is a Singapore exempt private company and wholly owned subsidiary of MindWave Innovations Inc (“TechyTrade (Singapore)”). TechyTrade FZ LLC is a limited liability company operating under the laws of the Ras Al Khaimah Economic Zone is a wholly owned subsidiary of TechyTrade Singapore (“TechyTrade (Dubai)”). MindWave operates through an international structure that includes TechyTrade (Singapore) and TechyTrade (Dubai), which contains the primary business operations of MindWave.
Our balance sheet strategy is designed to align with our DAT offering. TechyTrade (Dubai) owns or controls 1,000 Bitcoin free of encumbrances, identifiable and segregated within a sub-wallet architecture created and administered by MindWave Ltd. for the benefit of TechyTrade (Dubai), with private keys and beneficial ownership retained by TechyTrade (Dubai). TechyTrade (Dubai) has no indebtedness other than trade payables, which were less than fifty percent of cash on hand. This reserve posture is intended to preserve purchasing power, provide continuous exposure to Bitcoin, and support our yield infrastructure without requiring asset sales.
26
We intend to commercialize our principal DAT offerings for institutional clients, which comprise: (i) secure corporate Bitcoin treasury infrastructure designed for public-company balance sheets and institutional controls; (ii) AI-supported Bitcoin yield strategies intended to deliver risk-managed, programmatic yield including InsureTech; and (iii) a validator-enabled ecosystem that supports utility and governance via $NILA across interoperable verticals, including AdTech engagement platforms and ClimateTech impact systems, as described in our technical materials. Custody of client assets is expected to occur through regulated third-party providers and institutional-grade wallet solutions, implemented within a segregation and control framework consistent with institutional compliance requirements.
Our operating framework is designed around Bitcoin as the core reserve and risk collateral reference for treasury and yield solutions. We expect to avoid hybrid collateral models in favor of a Bitcoin-centric approach that is transparent, simple, and consistent with our treasury and platform design. Within our ecosystem, $NILA functions as an economic and governance substrate to activate services, enable staking mechanisms that align incentives, and facilitate validator economics across our interoperable platforms.
We intend to serve corporate treasuries and institutions that maintain or plan to maintain Bitcoin reserves and require scalable, compliant DAT capabilities. We also expect to support high-net-worth individuals and funds seeking institutional controls for Bitcoin reserve management and programmatic yield. Consistent with institutional practices, our anticipated operations emphasize transparency and risk management, including conservative collateralization where relevant to client programs, real-time risk monitoring, and robust anti-money laundering (AML) and know-your-customer (KYC) processes. We are pursuing strategic relationships with financial institutions, placement agents, and digital-asset service providers to support custody, treasury operations, and capital formation for expansion of our platform.
We believe that our combination of (i) an institutional Bitcoin reserve posture supported by segregated custody architecture and independent attestation, (ii) AI-enhanced yield strategies integrated into a controlled DAT stack, and (iii) a validator-enabled ecosystem powered by $NILA positions us to meet emerging institutional demand for Bitcoin treasury solutions. In connection with our merger with Apimeds Pharmaceuticals US, Inc., our DAT platform is expected to be operated within a publicly listed structure, with Apimeds’ biopharmaceutical business continuing within a subsidiary post-closing, and capital formation initiatives contemplated to support both the DAT platform and biopharmaceutical development. We believe this structure enhances our ability to scale client adoption while maintaining regulatory discipline and institutional-grade controls.
General Development of Our Business
MindWave Innovations Inc was incorporated in the State of Delaware in 2025. On December 1, 2025, MindWave signed and closed an Agreement and Plan of Merger with Apimeds Pharmaceuticals US, Inc. (“Apimeds”), Apimeds Merger Sub, Inc. (“Merger Sub”), Lokahi Therapeutics, Inc., and Erik Emerson, pursuant to which Merger Sub merged with and into MindWave, with MindWave surviving as a wholly owned subsidiary of Apimeds (the “Merger”).
Following the Merger, we have focused our development efforts on scaling an institutional DAT platform. Consistent with our treasury reserve posture, our group maintains Bitcoin as its core reserve asset. As previously mentioned, TechyTrade (Dubai) owns and controls 1,000 Bitcoin, identifiable and segregated in sub-wallets administered by MindWave Ltd.
Our principal activities consist of: (i) building our institutional DAT stack, including secure corporate Bitcoin treasury infrastructure and AI-supported Bitcoin yield strategies; (ii) implementing and attesting to our Bitcoin reserve and segregated custody architecture; and (iii) forming relationships with regulated custodians, wallet and validator providers, financial institutions, and placement agents to support custody, risk management, compliance (including AML/KYC), and capital formation. We are in the execution stage and are preparing for commercialization of our DAT offerings.
Roadmap and Milestones
Our near-term priorities focus on: (i) onboarding initial institutional clients to our corporate Bitcoin treasury infrastructure; (ii) progressing AI-supported, risk-managed yield programs under defined policy and control frameworks; (iii) standing up enterprise validator services to support network participation and governance; and (iv) advancing development of our ClimateTech, AdTech, and InsurTech verticals to interoperate with our DAT stack. Over time, we intend to expand treasury management capabilities, scale validator operations, and develop ecosystem utility, subject to applicable regulatory requirements, market conditions, and governance approvals.
27
Our Treasury Strategy and Digital Asset Treasury Platform
Bitcoin Treasury Strategy
We believe that Bitcoin is an attractive reserve asset because (i) it can serve as a store of value supported by a robust, public, open-source architecture that is independent of sovereign monetary policy, (ii) its fixed supply offers the potential to serve as a long-term hedge against inflation and, as adoption increases, the opportunity for appreciation, and (iii) the Bitcoin network provides infrastructure for financial and technological innovation.
We were formed with the intention of operating an institutional-focused Digital Asset Treasury (“DAT”) platform, and our group has adopted a Bitcoin-centric reserve posture aligned with that platform. Under this posture, our treasury reserve assets consist principally of:
| ● | Cash and cash equivalents sufficient for working capital and operational needs; and |
| ● | Bitcoin held at the operating-subsidiary level as our primary reserve asset, maintained within a segregated sub-wallet architecture and free of encumbrances, subject to business needs and market conditions. |
Consistent with this reserve posture, we may from time to time evaluate capital raising transactions to support platform development, working capital, and expansion of our reserve strategy. We expect future capital allocation decisions to consider market conditions, risk management, regulatory considerations, and anticipated operating requirements. Our strategy contemplates that we may (i) periodically rebalance or sell Bitcoin for general corporate purposes, (ii) utilize our Bitcoin reserve within conservative, risk-managed programs that support our yield and validator infrastructure, and (iii) evaluate compliant structures to generate programmatic returns consistent with institutional practices and applicable law.
We also plan to conduct advocacy and educational initiatives regarding institutional Bitcoin treasury standards, custody segregation, controls, and reporting, including thought leadership and partnerships intended to support the broader adoption of compliant corporate Bitcoin treasury operations.
DAT Yield and Validator Infrastructure
Our core offerings center on an institutional DAT stack designed to help corporations and institutions hold, manage, and generate yield on Bitcoin reserves. Key components include:
| ● | Institutional treasury infrastructure. Secure wallet architecture with segregation and controls designed to align with public-company balance sheet requirements and institutional compliance frameworks. |
| ● | AI-supported yield programs. Risk-managed strategies intended to produce programmatic returns on Bitcoin reserves, with governance and monitoring controls suitable for institutional participants. |
| ● | Validator-enabled ecosystem. Operations that support network economics and governance across our ecosystem, including utility powered by our native token, $NILA. |
Client assets, if and when onboarded, are expected to be held through regulated third-party custodians or institutional-grade wallet solutions within a segregation and control framework consistent with AML/KYC and other applicable requirements. We emphasize conservative collateralization (where relevant to client programs), real-time risk monitoring, and robust compliance processes.
Our Bitcoin Holdings
On March 31, 2025, TechyTrade (Dubai) entered into an Asset Purchase Agreement with MindWave Ltd., pursuant to which TechyTrade (Dubai) acquired 1,000 bitcoin. The bitcoin are held free of encumbrances within a segregated sub-wallet architecture administered by MindWave Ltd. We did not sell any bitcoin during 2024 or 2025.
28
Our Industry Overview
Bitcoin Industry
Bitcoin is a digital asset that is issued by and transmitted through an open-source protocol, known as the Bitcoin protocol, collectively maintained by a peer-to-peer network of decentralized user nodes. This network hosts a public transaction ledger, known as the Bitcoin blockchain, on which bitcoin holdings and all validated transactions that have ever taken place on the Bitcoin network are recorded. Balances of bitcoin are stored in individual “wallet” functions, which associate network public addresses with one or more “private keys” that control the transfer of bitcoin. The Bitcoin blockchain can be updated without any single entity owning or operating the network.
Creation of New Bitcoin and Limits on Supply
The Bitcoin protocol limits the total number of bitcoins that can be generated over time to 21 million. As of October 2025, approximately 19.93 million bitcoins have been generated. New bitcoins are created and allocated by the Bitcoin protocol through a “mining” process that rewards users that validate transactions in the Bitcoin blockchain. Validated transactions are added in “blocks” approximately every 10 minutes. The mining process serves to validate transactions and secure the Bitcoin network. Mining is a competitive and costly operation that requires a large amount of computational power to solve complex mathematical algorithms. This expenditure of computing power is known as “proof of work.”
To incentivize miners to incur the costs of mining bitcoin, the Bitcoin protocol rewards miners that successfully validate a block of transactions with newly generated bitcoin. The current reward for miners that successfully validate a block of transactions is 3.125 bitcoin per mined block. The mining reward is reduced by half, which is referred to as a bitcoin halving, after every 210,000 blocks are mined. This has historically occurred approximately every four years. The most recent bitcoin halving occurred in April 2024, and the next bitcoin halving is expected to occur sometime in 2028.
Modifications to the Bitcoin Protocol
Bitcoin is an open-source network that has no central authority, so no one person can unilaterally make changes to the software that runs the network. However, there is a core group of developers that maintains the code for the Bitcoin protocol, and they can propose changes to the source code and release periodic updates and other changes. Unlike most software that has a central entity that can push updates to users, bitcoin is a peer-to-peer network in which individual network participants, called nodes, decide whether to upgrade the software and accept the new changes. As a practical matter, a modification becomes part of the Bitcoin protocol only if the proposed changes are accepted by participants collectively having more than 50% of the processing power, known as hash rate, on the network. If a certain percentage of the nodes reject the changes, then a “fork” takes place, and participants can choose the version of the software they want to run.
Forms of Attack Against the Bitcoin Network and Wallets
Blockchain technology has many built-in security features that make it difficult for hackers and other malicious actors to corrupt the protocol or blockchain. However, as with any computer network, the Bitcoin network may be subject to certain attacks. Some forms of attack include unauthorized access to wallets that hold bitcoin and direct attacks, like “51% attacks” or “denial-of-service attacks” on the Bitcoin network.
Bitcoin is controllable only by the possessor of both the unique public key and private key(s) relating to the local or online digital wallet in which the bitcoin is held. Private keys used to access bitcoin balances are not widely distributed and are typically held on hardware (which can be physically controlled by the holder or by a third party such as a custodian) or via software programs on third-party servers. One form of obtaining unauthorized access to a wallet occurs following a phishing attack where the attacker deceives the victim and manipulates them into sharing their private keys for their digital wallet or other sensitive information. Other similar attacks may also result in the loss of private keys and the inability to access, and effective loss of, the corresponding bitcoin.
A “51% attack” may occur when a group of miners attain more than 50% of the Bitcoin network’s mining power, thereby enabling them to control the Bitcoin network and protocol and manipulate the blockchain. A “denial-of-service attack” occurs when legitimate users are unable to access information systems, devices, or other network resources due to the actions of a malicious actor flooding the network with traffic until the network is unable to respond or crashes. The Bitcoin network has been, and can be in the future, subject to denial-of-service attacks, which can result in temporary delays in block creation and in the transfer of bitcoin.
29
Bitcoin Industry Participants
The primary Bitcoin industry participants are miners, investors and traders, digital asset exchanges and service providers, including custodians, brokers, payment processors, wallet providers and financial institutions.
Miners. Miners range from bitcoin enthusiasts to professional mining operations that design and build dedicated mining machines and data centers, including mining pools, which are groups of miners that act cohesively and combine their processing power to mine bitcoin blocks. See “— Creation of New Bitcoin and Limits on Supply” above.
Investors and Traders. Bitcoin investors and traders include individuals and institutional investors who, directly or indirectly, purchase, hold, and sell bitcoin or bitcoin-based derivatives. On January 10, 2024, the Securities and Exchange Commission (“SEC”) issued an order approving several applications for the listing and trading of shares of spot bitcoin exchange-traded products (“ETPs”) on U.S. national securities exchanges. While the SEC had previously approved exchange-traded funds where the underlying assets were bitcoin futures contracts, this order represented the first time the SEC approved the listing and trading of ETPs that acquire, hold and sell bitcoin directly. ETPs can be bought and sold on a stock exchange like traditional stocks, and provide investors with another means of gaining economic exposure to bitcoin through traditional brokerage accounts.
Digital Asset Exchanges. Digital asset exchanges provide trading venues for purchases and sales of bitcoin in exchange for fiat or other digital assets. Bitcoin can be exchanged for fiat currencies, such as the U.S. dollar, at rates of exchange determined by market forces on bitcoin trading platforms, which are not regulated in the same manner as traditional securities exchanges. In addition to these platforms, over-the-counter markets and derivatives markets for bitcoin also exist. The value of bitcoin within the market is determined, in part, by the supply of and demand for bitcoin in the global bitcoin market, market expectations for the adoption of bitcoin as a store of value, the number of merchants that accept bitcoin as a form of payment, and the volume of peer-to-peer transactions, among other factors.
Service providers. Service providers offer a multitude of services to other participants in the Bitcoin industry, including custodial and trade execution services, commercial and retail payment processing, wallet solutions and financial institutions. If adoption of the Bitcoin network continues to materially increase, we anticipate that service providers may expand the currently available range of services and that additional parties will enter the service sector for the Bitcoin network.
Other Digital Assets
As of the date of this Annual Report, bitcoin was the largest digital asset by market capitalization. However, numerous alternative digital assets exist, and many entities, including consortia and financial institutions, are actively researching and investing resources in blockchain platforms and digital assets that utilize consensus mechanisms other than proof-of-work mining, which is employed by the Bitcoin network. For example, in late 2022, the Ethereum network transitioned to a “proof-of-stake” mechanism for validating transactions that requires significantly less computing power than proof-of-work mining. Other alternative digital assets that compete with bitcoin in certain ways include “stablecoins,” which are designed to maintain a constant price because of their issuers’ promise to hold high-quality liquid assets (such as U.S. dollar deposits and short-term U.S. treasury securities) equal to the total value of stablecoins in circulation. Stablecoins have grown rapidly as an alternative to bitcoin and other digital assets as a medium of exchange and store of value, particularly on digital asset trading platforms. As of December 31, 2024, two of the eight largest digital assets by market capitalization were U.S. dollar-backed stablecoins.
Additionally, central banks in some countries have started to introduce digital forms of legal tender. For example, China’s central bank digital currency (“CBDC”) project was made available to consumers in January 2022, and governments including the United States and the European Union have discussed the potential creation of new CBDCs.
30
Our Market Opportunity for Institutional Bitcoin Treasury and DAT Solutions
The use of Bitcoin as a corporate treasury reserve asset has expanded as companies and institutions evaluate alternatives to traditional cash management strategies. While fiat currency and short-term government securities remain predominant, the emergence of U.S. spot Bitcoin exchange-traded funds and increasing recognition of Bitcoin as an investable asset class have contributed to broader institutional acceptance. A growing number of public and private companies, investment funds, and other institutional allocators have begun to evaluate or adopt Bitcoin reserves as a potential long-term store of value and as a component of diversified treasury strategies.
Despite these developments, adoption of Bitcoin for treasury purposes remains limited relative to the overall size of global corporate treasury balances. We believe this gap highlights a meaningful market opportunity for institutional-grade platforms that can provide compliant, transparent, and risk-managed infrastructure for Bitcoin treasury operations. Key needs we see in the market include secure custody with segregation and controls; governance frameworks aligned with public-company standards; accounting and reporting support; risk-managed yield programs; and integration into existing treasury workflows.
We believe there is an opportunity to differentiate by offering an integrated DAT solution that combines: (i) corporate Bitcoin treasury infrastructure designed for institutional policies and controls; (ii) AI-supported, risk-managed yield programs intended to produce programmatic returns on reserve assets; and (iii) a validator-enabled ecosystem that supports network participation and governance, with utility powered by our native token, $NILA. As institutional comfort and regulatory clarity continue to develop, we expect the use of Bitcoin in treasury management to expand, supported by demand for transparent, regulated, and operationally disciplined solutions.
In our view, market growth will be driven by several secular trends: increased institutional access to Bitcoin through regulated vehicles and service providers; enhancements in custody, compliance, and accounting standards; and the need for end-to-end platforms that allow corporate treasury teams to implement Bitcoin strategies without building bespoke infrastructure. Our platform is being developed to address these requirements by providing segregation, controls, monitoring, and reporting consistent with institutional expectations, along with programmatic yield and validator capabilities designed to complement a Bitcoin-centric reserve posture.
Our Products and Services
Our principal planned products and services focus on delivering an institutional Digital Asset Treasury (“DAT”) platform that enables corporations and institutions to hold, manage, and generate yield on Bitcoin reserves through a compliant, scalable infrastructure. We are in the development execution stage and are actively building the underlying architecture, governance, and partnerships necessary to support future commercialization.
Institutional DAT Platform
| ● | Corporate Bitcoin treasury infrastructure. Segregated wallet architecture, permissions and controls, and reporting designed to align with institutional compliance frameworks and public-company balance sheet requirements. |
| ● | AI-supported yield programs. Risk-managed strategies intended to produce programmatic returns on Bitcoin reserves, with governance, monitoring, and risk limits tailored for institutional participants. |
| ● | Validator-enabled ecosystem. Network operations that support validator economics and governance across our interoperable platforms, with utility powered by our native token, $NILA. |
| ● | Compliance and risk management. Policies and tooling addressing AML/KYC, ongoing monitoring, collateral and reserve discipline where relevant to client programs, and real-time risk analytics. |
| ● | Advisory and enablement. Onboarding and operational support to help clients adopt institutional Bitcoin treasury standards, including segregation, reconciliation, and controls. |
Treasury Reserve and Bitcoin Strategy
Our group maintains a Bitcoin-centric reserve posture that aligns with our DAT platform. This approach is intended to preserve purchasing power, provide continuous exposure to Bitcoin, and support our yield and validator infrastructure. From time to time, we may evaluate capital raising transactions to fund platform development, operations, and reserve strategy, subject to market conditions, risk management, regulatory considerations, and anticipated operating needs.
31
Custody and Execution Services
We intend to utilize regulated third-party custodians and institutional-grade wallet solutions for the safekeeping of any client assets onboarded to our platform, as well as our operating-subsidiary reserves. These providers are expected to support secure custody, segregation, and controls, and to offer affiliated execution services for Bitcoin acquisitions and dispositions consistent with institutional best practices.
Ecosystem Verticals Powered by $NILA
In addition to our core Digital Asset Treasury (“DAT”) platform, we operate interoperable verticals powered by our native token, $NILA, which enables governance, utility, and value flow across our ecosystem.
| ● | ClimateTech (AQUAE Impact). One our ecosystems is a blockchain-powered sustainability engine intended to generate verifiable, insured environmental impact outcomes, including fractionalized credits linked to analog forest projects and other conservation efforts. The roadmap contemplates phases focused on expanding analog forest capacity, introducing water-conservation and biodiversity credits, and building a secondary market for the exchange of such credits. |
| ● | AdTech (Wave+). Another ecosystem is a micro-engagement platform to convert user attention into tokenized value for brand and mission-aligned campaigns. The platform is designed to support daily active engagement, with contemplated revenue streams from ad placements, partner campaigns, and conversion fees. |
| ● | InsurTech (Institutional Insurance Engine). We are designing an insurance framework to complement digital treasury strategies through surplus-capital-style models backed by strengthened, Bitcoin-centric balance sheets. The goal is to support treasury protection, parametric digital-asset coverage, and institutional insurance primitives that can be integrated with our DAT platform. |
Validator Infrastructure
We intend to operate enterprise-grade validator nodes to support network participation, governance, and rewards within our ecosystem and for institutional clients. The validator stack is being developed with an emphasis on high availability, slashing prevention, hardware security (including HSM/TEE-based protections), governance controls, and policy-limited MEV/PBS participation. Anticipated revenue drivers include staking rewards, protocol incentives, enterprise validator service fees, and ecosystem transaction fees.
Revenue Model
Our contemplated revenue model includes multiple streams that align with institutional treasury adoption:
| ● | Treasury Management Fees: Asset-under-management-based fees for corporate treasury wallets and related services. |
| ● | Performance and Program Fees: Participation in net returns from AI-supported yield programs, subject to institutional governance and risk policies. |
| ● | Validator Services and Protocol Incentives: Enterprise staking and validator service fees, protocol-derived incentives, and transaction-based revenues tied to ecosystem activity. Monetization associated with ClimateTech (e.g., fractionalized credits), AdTech (campaign and placement revenue), and InsurTech-related structures, where applicable. |
32
Our Customers
We target enterprise clients that require institutional governance, custody segregation, and audit-ready reporting for Bitcoin treasury operations, including public and private corporate treasury teams, institutional investors and asset managers, financial institutions and intermediaries, and high-net-worth organizations with significant Bitcoin reserves. We aim to integrate into CFO, treasury, and risk committee workflows, including policy design, compliance and accounting support, and board-level reporting.
We have generated revenues and do not currently rely on any single customer for more than 10% of our revenues.
Our Market Positioning
We believe our market positioning is strengthened by the integration of our institutional Digital Asset Treasury (“DAT”) platform, segregated custody architecture, and Bitcoin-centric reserve posture. Our platform is designed to enable corporations and institutions to implement Bitcoin treasury operations with the governance, controls, and reporting expected by institutional stakeholders, while complementing those reserves with AI-supported, risk-managed yield programs and validator participation within our broader ecosystem.
Our Bitcoin holdings are maintained primarily as a treasury reserve asset intended to support our balance sheet and operational resilience. Our reserve posture reinforces our credibility with institutional clients by aligning our incentives with a Bitcoin-centric treasury strategy and by demonstrating disciplined custody, segregation, and control practices.
By maintaining a Bitcoin-centric reserve posture and building end-to-end DAT infrastructure, we aim to provide a comprehensive, compliant, and scalable pathway for institutions to adopt Bitcoin in treasury management. This integration of reserve management, custody controls, yield programs, and validator operations is intended to enhance risk management, support operational discipline, and provide a foundation for long-term value creation.
Our Competitive Strengths
Platform Governance and $NILA Utility
$NILA functions as a governance and utility token within our ecosystem, facilitating validator participation, program access, and value exchange across verticals. Governance processes are designed to be policy-driven and auditable, with the objective of aligning incentives among stakeholders while maintaining institutional controls. Within our validator framework, we intend to limit protocol participation to policy-permitted activities and to maintain risk controls, including stake diversification, slashing protection, and real-time monitoring.
We believe the following strengths position us to compete effectively in the emerging institutional Bitcoin treasury market:
| ● | Institutional DAT stack. End-to-end infrastructure for corporate Bitcoin treasury operations, including secure wallet architecture, permissions and controls, reconciliation, and reporting aligned with institutional requirements. |
| ● | Segregated custody architecture. Use of institutional-grade wallet solutions and regulated third-party providers for safekeeping, with segregation and control frameworks designed to mitigate counterparty and operational risks. |
| ● | Bitcoin-centric reserve posture. A treasury strategy centered on Bitcoin reserves intended to preserve purchasing power, maintain continuous exposure, and support platform credibility with institutional clients. |
| ● | AI-supported yield programs. Risk-managed strategies intended to produce programmatic returns on reserve assets, governed by policies, monitoring, and limits consistent with institutional risk management. |
| ● | Validator-enabled ecosystem. Network participation that supports validator economics and governance across interoperable platforms, with utility powered by our native token, $NILA. |
| ● | Compliance and governance. Emphasis on AML/KYC, surveillance, and real-time risk monitoring, together with operational controls designed to align with institutional and public-company standards. |
| ● | Scalable partner network. Strategic relationships with custodians, wallet providers, financial institutions, and other service partners to support onboarding, execution, and operational scale. |
| ● | Operational discipline. A focus on segregation, transparency, and audit-ready processes to support institutional adoption and long-term sustainability. |
33
We believe our integrated approach — combining corporate Bitcoin treasury infrastructure, AI-supported yield programs, validator operations, and interoperable verticals powered by $NILA — addresses key adoption barriers by offering a policy-driven, audit-ready, and scalable pathway for institutions. Our multi-vertical design is intended to enhance network effects and create diversified revenue opportunities aligned with institutional compliance and governance expectations.
Custody of Our Bitcoin
Our Bitcoin reserve is maintained within a segregated sub-wallet architecture created and administered by MindWave Ltd. for the benefit of our operating subsidiary, TechyTrade (Dubai). TechyTrade (Dubai) holds the private keys and retains full beneficial ownership and control of the segregated wallet(s). The 1,000 Bitcoin are identifiable, segregated, and held free and clear of any encumbrances. This structure is intended to mitigate counterparty exposure associated with omnibus custody while maintaining operational controls and traceability over our reserve assets.
We conduct diligence and ongoing oversight of our wallet architecture and related service providers, focusing on segregation, key-management controls, access permissions, and incident-response procedures. Our controls emphasize least-privilege access, multi-factor authentication, operational separation of duties, and audit trails for key interactions and movement authorizations. We supplement these controls with periodic independent attestations to verify the existence, segregation, and control of our Bitcoin holdings as of specified dates.
For any client assets that may be onboarded to our platform in the future, we expect to utilize regulated third-party custodians and institutional-grade wallet solutions, implemented within a segregation and control framework designed to meet institutional compliance requirements (including AML/KYC, monitoring, and reporting). We anticipate diversifying custody solutions as appropriate to support risk management and operational resilience, and we will evaluate additional custodial partners over time.
Where we engage third-party providers, we expect to conduct initial and periodic reviews of their security, operations, and controls, including — but not limited to — key-management practices, cold-storage policies, cybersecurity programs, business continuity and disaster recovery readiness, and the availability of third-party assurance reporting. We also seek contractual provisions addressing segregation of assets and clarifying that digital assets held for our benefit are not part of a custodian’s bankruptcy estate under applicable law.
We continuously monitor the safekeeping of our Bitcoin through internal controls, reconciliation, and external confirmations, and we will conduct supplemental diligence when warranted by market conditions or other circumstances.
Policy Framework and Risk Management
We are building our platform around policy-based governance intended to satisfy institutional requirements and auditor expectations. Core elements include board-approved asset allocation and transaction policies, segregation of duties, multi-step approval workflows, transaction screening, real-time solvency and exposure limits, and emergency protocol “kill-switches.” For yield programs, we intend to maintain leverage caps, counterparty concentration limits, daily reconciliations, and tail-risk hedging parameters. For validator operations, we emphasize uptime targets, redundancy, slashing prevention, and pre-defined MEV/PBS participation rules. We expect to produce periodic “audit packs,” including reconciliations and control attestations, for institutional users.
Potential Advantages and Disadvantages of Holding Bitcoin
We believe that bitcoin is an attractive asset because it can serve as a store of value, supported by a robust and public open-source architecture, that is untethered to sovereign monetary policy. We also believe that, due to its limited supply, bitcoin offers the potential to serve as a hedge against inflation in the long-term and, if its adoption increases, the opportunity for appreciation in value.
34
Bitcoin exists entirely in electronic form, as virtually irreversible public transaction ledger entries on the blockchain, and transactions in bitcoin are recorded and authenticated not by a central repository, but by a decentralized peer-to-peer network. This decentralization mitigates the risks of certain threats common to centralized computer networks, such as denial-of-service attacks, and reduces the dependency of the bitcoin network on any single system. The decentralization of user nodes and miners also mitigates the risk of a 51% attack, which would be very costly and difficult to execute with respect to bitcoin because the Bitcoin network is open source and widely distributed, and transactions on the blockchain require significant computing power to be validated. However, while the Bitcoin network as a whole is decentralized, the private keys used to access bitcoin balances are not widely distributed and are susceptible to phishing and other attacks designed to obtain sensitive information or gain access to password-protected systems. Loss of such private keys can result in an inability to access, and effective loss of, the corresponding bitcoin. Consequently, bitcoin holdings are susceptible to all of the risks inherent in holding any electronic data, such as power failure, data corruption, security breach, communication failure and user error, among others. These risks, in turn, make bitcoin substantially more susceptible to theft, destruction, or loss of value from hackers, corruption, viruses and other technology-specific factors as compared to conventional fiat currency or other conventional financial assets.
In addition, the Bitcoin network relies on open-source developers to maintain and improve the Bitcoin protocol. Accordingly, bitcoin may be subject to protocol design changes, governance disputes such as “forked” protocols, competing protocols, and other open source-specific risks that do not affect conventional proprietary software.
Government Regulation
The laws and regulations applicable to bitcoin and digital assets are evolving and subject to interpretation and change.
Governments around the world have reacted differently to digital assets; certain governments have deemed them illegal, and others have allowed their use and trade without restriction, while in some jurisdictions, such as the U.S., digital assets are subject to overlapping, uncertain and evolving regulatory requirements.
As digital assets have grown in both popularity and market size, the U.S. Executive Branch, Congress and a number of U.S. federal and state agencies, including the Financial Crimes Enforcement Network (“FinCEN”), the Commodity Futures Trading Commission (“CFTC”), the SEC, the Financial Industry Regulatory Authority (“FINRA”), the Consumer Financial Protection Bureau (“CFPB”), the Department of Justice, the Department of Homeland Security, the Federal Bureau of Investigation, the Internal Revenue Service (“IRS”) and state financial regulators, have been examining the operations of digital asset networks, digital asset users and digital asset exchanges, with particular focus on the extent to which digital assets can be used to violate state or federal laws, including to facilitate the laundering of proceeds of illegal activities or the funding of criminal or terrorist enterprises, and the safety and soundness and consumer-protective safeguards of exchanges or other service-providers that hold, transfer, trade or exchange digital assets for users. Many of these state and federal agencies have issued consumer advisories regarding the risks posed by digital assets to investors. In addition, federal and state agencies, and other countries have issued rules or guidance regarding the treatment of digital asset transactions and requirements for businesses engaged in activities related to digital assets.
Depending on the regulatory characterization of bitcoin, the markets for bitcoin in general, and our activities in particular, our business and our bitcoin strategy may be subject to regulation by one or more regulators in the United States and globally. Ongoing and future regulatory actions may alter, to a materially adverse extent, the nature of digital assets markets, the participation of industry participants, including service providers and financial institutions in these markets, and our ability to pursue our bitcoin strategy. Additionally, U.S. state and federal and foreign regulators and legislatures have taken action against industry participants, including digital assets businesses, and enacted restrictive regimes in response to adverse publicity arising from hacks, consumer harm, or criminal activity stemming from digital assets activity. U.S. federal and state energy regulatory authorities are also monitoring the total electricity consumption of cryptocurrency mining, and the potential impacts of cryptocurrency mining to the supply and dispatch functionality of the wholesale grid and retail distribution systems. Many state legislative bodies have passed, or are actively considering, legislation to address the impact of cryptocurrency mining in their respective states.
The CFTC takes the position that some digital assets, including bitcoin, fall within the definition of a “commodity” under the Commodities Exchange Act of 1936, as amended (the “CEA”). Under the CEA, the CFTC has broad enforcement authority to police market manipulation and fraud in spot digital assets markets in which we may transact. Beyond instances of fraud or manipulation, the CFTC generally does not oversee cash or spot market exchanges or transactions involving digital asset commodities that do not utilize margin, leverage, or financing. In addition, CFTC regulations and CFTC oversight and enforcement authority apply with respect to futures, swaps, other derivative products and certain retail leveraged commodity transactions involving digital asset commodities, including the markets on which these products trade.
35
The SEC and its staff have taken the position that certain other digital assets fall within the definition of a “security” under the U.S. federal securities laws. Public statements made by senior officials and senior members of the staff at the SEC indicate that the SEC does not consider bitcoin to be a security under the federal securities laws. However, such statements are not official policy statements by the SEC and reflect only the speakers’ views, which are not binding on the SEC or any other agency or court and cannot be generalized to any other digital assets. In addition, since transactions in bitcoin provide a degree of anonymity, they are susceptible to misuse for criminal activities, such as money laundering. This misuse, or the perception of such misuse, could lead to greater regulatory oversight of bitcoin and Bitcoin platforms, and there is the possibility that law enforcement agencies could close or blacklist bitcoin platforms or other bitcoin-related infrastructure with little or no notice and prevent users from accessing or retrieving bitcoin held via such platforms or infrastructure. For example, the U.S. Treasury Department’s Office of Foreign Assets Control has issued updated advisories regarding the use of virtual currencies, added a number of digital asset exchanges and service providers to the Specially Designated Nationals and Blocked Persons list and engaged in several enforcement actions, including a series of enforcement actions that have either shut down or significantly curtailed the operations of several smaller digital asset exchanges associated with Russian and/or North Korean nationals. Additionally, in January 2025, the Consumer Financial Protection Bureau announced that it is seeking public input on privacy protections and surveillance in digital payments, particularly those offered through large technology platforms.
As noted above, activities involving bitcoin and other digital assets may fall within the jurisdiction of more than one financial regulator and various courts and such laws and regulations are rapidly evolving and increasing in scope. On January 23, 2025, President Trump issued an executive order titled, Strengthening American Leadership in Digital Financial Technology. While the executive order did not mandate the adoption of any specific regulations, the executive order identifies certain key objectives to guide agencies involved in crypto regulation, including (i) protecting the sovereignty of the United States dollar by promoting the development of United States dollar-backed stablecoins, (ii) providing regulatory clarity and certainty built on technology-neutral regulations for individuals and firms involved in digital assets, including through well-defined jurisdictional regulatory boundaries, and (iii) taking measures to protect Americans from the risks of Central Bank Digital Currencies. To achieve these objectives, the executive order established a working group on digital asset markets within the National Economic Council, comprised of representatives from key federal agencies, with a tight timeline for examining existing regulations and proposing a new regulatory framework. There have also been several bills introduced in Congress that propose to establish additional regulation and oversight of the digital asset markets.
Aspects of our business involve collecting, processing, disclosing, storing, and transmitting personal data, which are subject to certain privacy policies, contractual obligations, and U.S. and foreign laws, regulations, and directives relating to privacy and data protection.
There are a broad variety of other data protection laws in the United States that are or may be applicable to our activities, and a wide range of enforcement agencies at both the state and federal levels that can review companies for privacy and data security concerns based on general consumer protection laws. The Federal Trade Commission and state Attorneys General all are aggressive in reviewing privacy and data security protections for consumers. New laws also are being considered at both the state and federal levels. A broad range of legislative measures also have been introduced at the federal level. Accordingly, failure to comply with federal and state laws (both those currently in effect and future legislation) regarding privacy and security of personal information could expose us to fines and penalties under such laws. In the event of a security breach, we also may have obligations to notify our customers or other parties or individuals about this breach, and this can lead to significant costs and the risk of potential enforcement and/or litigation. There is also a threat of consumer class actions related to these laws and the overall protection of personal data. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which could harm our reputation and our business.
There are similar laws in other countries, including the General Data Protection Regulation (“GDPR”) in the European Union which imposes requirements regarding the handling and security of personal data, requires disclosure of data breaches to individuals, customers, and data protection authorities in certain circumstances, requires companies to honor data subjects’ requests relating to their personal data, permits regulators to impose fines of up to €20,000,000 or 4% of global annual revenue, whichever is higher, and establishes a private right of action.
36
In addition to these specific laws, we also are subject to other privacy, security, and data protection laws around the world. In addition to the laws in place already, other countries are also considering new or expanded laws governing privacy and data security that may impact our business practices. These laws may impact our ongoing business activities and our relationships with our business partners, customers and service providers.
Furthermore, the U.S. Congress is considering comprehensive privacy legislation. At this time, it is unclear whether Congress will pass such a law and if so, when and what it will require and prohibit. Moreover, it is not clear whether any such legislation would give the Federal Trade Commission (“FTC”) any new authority to impose civil penalties for violations of the Federal Trade Commission Act in the first instance, whether Congress will grant the FTC rulemaking authority over privacy and information security, or whether Congress will vest some or all privacy and data security regulatory authority and enforcement power in a new agency, akin to EU data protection authorities.
Sales and Marketing
Our sales and marketing strategy is designed to expand awareness of our institutional DAT platform, build credibility with corporate treasury teams and institutional allocators, and drive adoption of our offerings across target enterprise markets. Key elements include strategic partnerships, targeted distribution through enterprise sales and channels, and education-led marketing that emphasizes governance, compliance, and risk management.
Strategic Partnerships
We intend to rely on strategic relationships with regulated custodians, institutional-grade wallet providers, financial institutions, accounting and audit advisors, and other digital-asset service providers to support our treasury operations and client enablement. Because we are not directly licensed to provide custody, execution, or intermediation services, we expect these partners to supply the necessary regulatory infrastructure and, in certain cases, act as distribution channels. These relationships are expected to support secure custody, segregation and control frameworks, execution services for Bitcoin acquisitions and dispositions, validator operations, and programmatic risk management. We also expect to collaborate with placement agents and traditional financial institutions to broaden institutional reach and facilitate client onboarding. We will continue to evaluate additional partnerships with enterprise software integrators, cloud and security vendors, and protocol-level partners to enhance scale, resilience, and interoperability.
Distribution Methods
We plan to distribute our DAT solutions directly to enterprise clients through internal business development and relationship management teams focused on corporate treasuries, institutions, and financial intermediaries. In addition, we expect to leverage partnerships with custodians, wallet providers, and financial institutions to support client acquisition, onboarding, and ongoing operations. Over time, we anticipate supplementing these channels with participation in industry conferences and education-led initiatives, including research and thought-leadership publications tailored for CFOs, treasury leaders, boards, and risk committees. We do not expect to rely on mass-market retail marketing programs.
Marketing
We intend to emphasize education, transparency, and strategic engagement aligned with institutional standards. We expect to target the following principal audiences:
| ● | Corporate treasury managers and CFO organizations seeking to integrate Bitcoin into reserve strategies with institutional governance, controls, and reporting. |
| ● | Institutional investors and asset managers, including hedge funds and family offices, seeking institutional-grade custody, risk-managed yield programs, and validator-enabled capabilities. |
| ● | Financial institutions and intermediaries evaluating Bitcoin treasury offerings for their clients and seeking compliant, scalable infrastructure. |
| ● | High-net-worth individuals with significant Bitcoin holdings who require institutional-grade custody, governance, and programmatic operations. |
37
Our marketing strategy is expected to combine targeted enterprise outreach, channel partnerships, and education-driven content intended to promote responsible adoption of institutional Bitcoin treasury practices while reinforcing MindWave’s commitment to compliance, governance, and long-term value creation.
Intellectual Property
We rely on a combination of proprietary software, trade secrets, and contractual protections to develop and operate our DAT platform, including components related to wallet orchestration, segregation and controls, risk analytics, AI-supported yield programs, and validator operations. We also rely on confidentiality and invention-assignment agreements with employees, contractors, and partners. From time to time, we may pursue trademark protection for brand names and logos associated with our products and ecosystem (including, for example, MindWave and $NILA), and we will evaluate additional intellectual property protections as our platform develops and markets evolve.
Seasonality
Our business is not subject to material seasonal variations. However, our results of operations may be affected by fluctuations in the market price of bitcoin and by the timing of capital raising activities, which may not occur evenly throughout the year.
Employees
As of January 11, 2026, we had a total of 9 employees, none of whom are based in the United States. None of our employees are represented by a labor union. We have not experienced any work stoppages and generally consider our relations with our employees to be good.
Legal Proceedings
From time to time, we may be involved in legal proceedings, claims, or regulatory matters arising in the ordinary course of business. As of the date of this Annual Report, we are not a party to any material pending legal proceedings, nor are we aware of any such proceedings contemplated by governmental authorities.
On April 24, 2026, the Company, MindWave, and Lokahi Therapeutics, Inc., a Nevada corporation (“Lokahi” and, together with the Company and MindWave, the “Company Parties”), together with Erik Emerson, individually and in his capacity as Bio Business Representative under the Merger Agreement, entered into a Confidential Settlement and Mutual Release Agreement (the “Settlement Agreement”) with Inscobee Inc., a South Korean corporation (“Inscobee”), and Apimeds Inc., a South Korean corporation and wholly owned subsidiary of Inscobee (“Apimeds Korea” and, together with Inscobee, the “Inscobee Parties”). Concurrently with the Settlement Agreement, the Company Parties and the Inscobee Parties also entered into a Side Letter Agreement (Merger Unwind Conditions) (the “Side Letter”), which is incorporated into and forms part of the Settlement Agreement.
The Settlement Agreement resolves all outstanding disputes among the parties arising from the Merger Agreement and related transactions.
Also on April 30, 2026, the Company entered into a Forbearance Agreement (the “Forbearance Agreement”) with Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B (the “Investor”), which holds a senior convertible note in the aggregate original principal amount of $11,000,000 (the “Existing Note”), issued pursuant to a Securities Purchase Agreement, dated December 1, 2025 (the “Securities Purchase Agreement”).
Pursuant to the Forbearance Agreement, the Investor has agreed to forbear from exercising any of its rights or remedies under the Existing Note with respect to certain existing events of default (collectively, the “Existing Defaults”) during the period commencing on the date of the Forbearance Agreement through and including June 30, 2026 (or such later date as the Investor may elect in its sole discretion) (the “Forbearance Period”).
The Settlement Agreement, the Side Letter, and the Forbearance Agreement are described in the Company’s Current Report on Form 8-K filed with the SEC on May 3, 2026, and are incorporated herein by reference.
Facilities
Our principal executive offices are located at 60 Paya Lebar Road #04-23, Singapore 409051.
Available Information
Our website is located at www.mindwavedao.com.
Item 1A. Risk Factors
As a smaller reporting company, as defined in Rule 12b-2 of the Exchange Act, we are not required to provide the information required by this Item.
Item 1B. Unresolved Staff Comments
Not applicable.
38
Item 1C. Cybersecurity
Risk Management and Strategy
Managing Material Risks & Integrated Overall Risk Management
In the normal course of business, we may collect and store certain sensitive company information, including proprietary and confidential business information. We maintain various protections designed to safeguard against cyberattacks, including firewalls, key-based authentication, and virtual private networks. We protect against business interruption by backing up our major systems. We consider these cybersecurity risk management efforts as part of our broader risk management framework. This integration helps ensure that cybersecurity considerations are a fundamental part of our decision-making processes. Our management team works with our audit committee and board of directors (the “Board”) to evaluate and address cybersecurity risks in alignment with our business objectives and operational needs.
Engage Third Parties on Risk Management
To date, we have
Oversee Third Party Risk
We utilize various third-party software applications
in the functioning of our core business. We consider the cybersecurity practices of our
Risks from Cybersecurity Threats
We face risk from cybersecurity threats that could have a material adverse effect on our business, financial condition, results of operations, cash flows or reputation.
To date, we have not experienced any previous cybersecurity
incidents that have materially affected or are reasonably likely to
Governance
Board of Directors Oversight
Our Board is aware of the critical nature of managing risks associated with cybersecurity threats, and recognizes the significance of these threats to our operational integrity and stockholder confidence.
39
Risk Management Personnel
The audit committee is central to the Board’s
Management’s Role Managing Risk and Reporting to the Board
We do not currently have an employee who has significant and demonstrated professional IT management experience and possesses the requisite education, skills and experience needed to develop and execute our cybersecurity strategies. Presently, our senior management is responsible for monitoring our cybersecurity risks and maintaining an ongoing dialogue with the audit committee regarding emerging or potential cybersecurity risks as needed. The relationship between senior management and the audit committee regarding current and emerging cybersecurity concerns helps to integrate cybersecurity consideration into the Company’s broader strategic objectives.
Item 2. Properties
Our principal executive offices are located at 100 Matawan Rd, Suite 325, Matawan, New Jersey 07747. Our Digital Asset Treasury business operations are headquartered at 60 Paya Lebar Road #04-23, Singapore 409051.
We do not own any real property. We believe our current facilities are suitable for our current operations.
Item 3. Legal Proceedings
From time to time, we may be involved in legal proceedings, claims, or regulatory matters arising in the ordinary course of business. As of the date of this Annual Report, we are not a party to any material pending legal proceedings, nor are we aware of any such proceedings contemplated by governmental authorities.
Item 4. Mine Safety Disclosures
Not applicable.
40
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities Market Information
Our shares of common stock are traded on the NYSE American under the symbol “APUS”.
Holders
As of the date of this Annual Report, there were approximately 10 holders of record of our common stock.
Dividends
We have not paid any cash dividends on our common stock to date. It is the present intention of our Board to retain all earnings, if any, for use in our business operations and, accordingly, our Board does not anticipate declaring any dividends in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report.
Recent Sales of Unregistered Securities
On December 1, 2025, in connection with the Merger, we issued an aggregate of 7,477,017 shares of preferred stock to former stockholders of MindWave Innovations Inc. in exchange for all of the outstanding shares of MindWave common stock. These shares were issued in reliance upon the exemption from registration provided by Section 4(a)(2) of the Securities Act of 1933, as amended.
Item 6. [Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Overview
Apimeds Pharmaceuticals US, Inc. (“APUS,” the “Company,” “we,” “us,” or “our”) is a development-stage biopharmaceutical company incorporated in the State of Delaware. Our primary focus is the clinical development of Apitox, a purified honeybee venom-based drug candidate being evaluated for the treatment of acute pain and inflammation associated with knee osteoarthritis. We operate our biopharmaceutical business through our wholly owned subsidiary, Lokahi Therapeutics Inc. (“Lokahi”).
Fiscal year 2025 was a transformative year for the Company. In addition to advancing our biopharmaceutical pipeline and completing our initial public offering on the New York Stock Exchange on May 12, 2025, we completed a merger with MindWave Innovations Inc. (“MindWave”) on December 1, 2025. As a result of the Merger, the Company became a dual-segment operating company, adding a digital asset operations segment alongside our core biopharmaceutical business. MindWave, now a wholly owned subsidiary, operates the MindWaveDAO blockchain ecosystem and holds digital assets consisting of Bitcoin (“BTC”), Tether (“USDT”), and the MindWaveDAO native utility token (“NILA Tokens”). The Merger fundamentally changed the scale and composition of our balance sheet, adding approximately $145.3 million in identifiable net assets, primarily digital assets, and was effected through the issuance of 7,477,017 shares of Series A Convertible Preferred Stock.
We have not yet generated revenue from our biopharmaceutical operations and MindWave operations and do not expect to do so until we have successfully advanced Apitox through clinical development and commercialization. Our ability to continue as a going concern depends on our ability to raise additional capital and execute our operational plans, as discussed further under “Liquidity and Capital Resources” below.
41
Key Developments in Fiscal Year 2025
Initial Public Offering
On May 12, 2025, we completed our IPO on the New York Stock Exchange, issuing 3,375,000 shares of common stock and generating net proceeds of approximately $11.6 million after deducting underwriting discounts, commissions, and offering costs.
Business Combination with MindWave Innovations Inc.
On December 1, 2025, we completed the Merger with MindWave, which became a wholly owned subsidiary of the Company. The transaction was effected as a non-cash business combination, with consideration consisting solely of 7,477,017 shares of Series A Convertible Preferred Stock with an aggregate fair value of approximately $145.4 million, equivalent to the fair value of the net identifiable assets acquired. No goodwill was recorded. Through the Merger, we acquired digital assets with an aggregate fair value of approximately $146.3 million at the acquisition date, consisting of BTC ($90.8 million), USDT ($4.7 million), and NILA Tokens ($50.8 million), as well as the operations of the MindWaveDAO blockchain.
PIPE Convertible Note Financing
In connection with the Merger, on December 1, 2025, we entered into a Securities Purchase Agreement providing for the issuance of senior secured convertible notes in an aggregate maximum principal amount of $129 million, to be drawn in tranches at our election. On December 8, 2025, we executed the first tranche, issuing a senior secured convertible note with a principal amount of $10.9 million and gross proceeds of $10 million. Of the proceeds, $8 million is currently held in a Deposit Account Control Agreement (“DACA”) account, and $1.1 million was disbursed to MindWave to fund its operations.
Biopharmaceutical Development Activity
During 2025, we significantly increased development activity related to Apitox, incurring research and development expenses of $1.6 million. We also entered into an agreement with Prevail InfoWorks Inc., our Clinical Research Organization (“CRO”), establishing a prepaid balance of approximately $2 million to be applied against future clinical trial execution.
Results of Operations
Year Ended December 31, 2025 Compared to Year Ended December 31, 2024
(a) Revenue
We generated no revenue from biopharmaceutical operations during the years ended December 31, 2025 or 2024. Proceeds from the sale of NILA Tokens are recognized as realized gains on digital assets and are not classified as revenue under ASC 606.
42
(b) Research and Development Expenses
Research and development expenses were $1.6 million for the year ended December 31, 2025, compared to zero for the year ended December 31, 2024. The increase reflects the commencement of meaningful development activity related to Apitox during 2025, including consulting fees, manufacturing costs, and CRO expenses incurred in connection with our clinical development program.
(c) General and Administrative Expenses
General and administrative expenses increased to $10.2 million for the year ended December 31, 2025, from $1.3 million for the year ended December 31, 2024, an increase of approximately $8.9 million. The increase was driven by a significant expansion of corporate activity and headcount in connection with becoming a public company and completing the Merger. Key drivers included increased consulting and professional fees, payroll and benefits, stock-based compensation of approximately $2.0 million (consisting of option grants, common stock grants, and advisory warrants), merger and acquisition related fees, office rent and supplies, and travel expenses.
(d) Total Operating Expenses
Total operating expenses were $11.9 million for the year ended December 31, 2025, compared to $1.3 million for the year ended December 31, 2024, reflecting the significant increase in both R&D and G&A activity described above.
(e) Other Income (Expense)
Total other income was $5.9 million for the year ended December 31, 2025, compared to other expense of approximately $0.1 million for the year ended December 31, 2024. The improvement was driven mostly by the digital asset activities acquired through the MindWave Merger, which closed on December 1, 2025, and was reflected in our consolidated results for the period December 1 through December 31, 2025 only. Key components of other income (expense) are as follows:
| ● | Unrealized gain on digital assets of $1.8 million, reflecting fair value increases in BTC and NILA Token holdings between the acquisition date and December 31, 2025, partially offset by a decline in BTC value during the period. |
| ● | Realized gain on sale of digital assets of $4.2 million, reflecting gains recognized on the sale of approximately 4.3 million NILA Tokens for aggregate proceeds of approximately $1.8 million in USDT during the period December 1 through December 31, 2025. These proceeds are noncash, delivered in the form of USDT, described above. These proceeds are used to maintain liquidity as well as satisfy operational expenses directly pertaining to blockchain and token management. |
| ● | Trading gains, net of approximately $97, representing the net of market-making income and expenses associated with the maintenance and trading activity of the MindWaveDAO NILA token. These gains are non-cash in nature. |
| ● | Change in fair value of derivative liability of $55,146, reflecting the remeasurement of the derivative liability bifurcated from the PIPE convertible note at issuance on December 8, 2025 through December 31, 2025. The derivative arises from the variable conversion terms of the note, which allow conversion at a 20% discount to the minimum volume-weighted average price over a five-day lookback period. |
| ● | Change in fair value of warrant liability of $22,377, reflecting the remeasurement of the advisory warrant liability between issuance and reclassification to equity on August 5, 2025. |
| ● | Interest income of $0.1 million, earned primarily on IPO proceeds held in a money market account at PNC Bank. |
| ● | Interest expense of $0.3 million, consisting primarily of accretion of the discount attributable to the PIPE convertible note issued December 8, 2025, as well as accrued interest on the related party notes payable aggregating $500,100 in principal as of December 31, 2025. |
43
(f) Net Loss
Net loss was $6.0 million for the year ended December 31, 2025, compared to a net loss of $1.4 million for the year ended December 31, 2024. The increase reflects the scale-up of operations in connection with the IPO, Merger, and advancement of the Apitox development program, partially offset by digital asset gains recognized following the MindWave acquisition.
Net loss per common share, basic and diluted, was $(0.55) for the year ended December 31, 2025, based on weighted-average shares outstanding of 10,881,907. All potentially dilutive securities, including 149.5 million common share equivalents underlying the Series A Convertible Preferred Stock, 8.3 million shares underlying the PIPE convertible note, 1.1 million warrant shares, and 0.5 million stock option shares, were excluded from the diluted calculation as their inclusion would have been anti-dilutive.
Liquidity and Capital Resources
Overview
As of December 31, 2025, we had cash and cash equivalents of $1.6 million and restricted cash of $8.0 million, for total cash, cash equivalents, and restricted cash of $9.6 million. We also held short-term investments of $2.0 million, representing a certificate of deposit established by Lokahi to earn interest on funds not deployed in operations. Working capital, excluding the fair value of digital assets (which are subject to price volatility and liquidity risk and therefore excluded from management’s assessment of near-term operational liquidity), was approximately $3.2 million as of December 31, 2025.
The $8.0 million of restricted cash represents PIPE convertible note proceeds currently held in a DACA account, pending release upon conversion of the Series A Convertible Preferred Stock in accordance with the terms of the note agreement. These funds are not available for general operating purposes until released from the DACA.
Cash Flows
(a) Operating Activities
Net cash used in operating activities was $8.9 million for the year ended December 31, 2025, compared to $0.7 million for the year ended December 31, 2024. The increase reflects the significant expansion of operating activity in connection with the IPO, Merger, and clinical development programs. Non-cash charges included in operating activities consisted primarily of stock-based compensation of $3.2 million (inclusive of option grants, common stock grants, and advisory warrants), accretion of debt discount of $0.3 million, and non-cash digital asset operating expenses of $2.4 million, partially offset by unrealized and realized gains on digital assets.
(b) Investing Activities
Net cash used in investing activities was $10.0 million for the year ended December 31, 2025, consisting primarily of the $8.0 million deposit into the DACA restricted cash account, $2.0 million invested in short-term instruments, and $57,333 in purchases of property and equipment (consisting of computer equipment and furniture), partially offset by $15,345 of cash acquired in the Merger.
(c) Financing Activities
Net cash provided by financing activities was $20.6 million for the year ended December 31, 2025, consisting primarily of $11.6 million in net proceeds from the IPO, $10.0 million in gross proceeds from the PIPE convertible note, and $250,100 in proceeds from related party promissory notes, partially offset by $946,000 in cash issuance costs and $1.1 million disbursed to MindWave from PIPE proceeds.
44
Future Capital Requirements and Going Concern
Since inception, we have incurred recurring operating losses and negative cash flows from operations, and we have not generated revenue from our biopharmaceutical operations. For the year ended December 31, 2025, we reported a net loss of $6.0 million and used $8.9 million in cash in operating activities. As of December 31, 2025, our accumulated deficit was $10.3 million. We expect to continue to incur significant operating losses as we advance the clinical development of Apitox and build our operational infrastructure. Significant manufacturing costs related to Apitox development are expected to be incurred beginning in the first quarter of 2026 and throughout the year. These conditions raise substantial doubt about our ability to continue as a going concern.
Management’s primary plan to address near-term liquidity needs is the continued execution of additional tranches under the Securities Purchase Agreement, which provides for the issuance of up to $129.0 million in aggregate principal of senior secured convertible notes. As of December 31, 2025, only the first tranche of $10.9 million in principal ($10.0 million gross proceeds) has been drawn, leaving substantial capacity available under the facility. Management believes that the remaining available tranches under the Securities Purchase Agreement, combined with continued NILA Token sales for USDT proceeds and potential additional equity or debt financing, represent the primary sources of capital to fund operations over the next twelve months. However, there can be no assurance that future tranches will be drawn, that NILA Token sales will generate sufficient proceeds, or that additional financing will be available on acceptable terms or at all. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Contractual Obligations and Commitments
Our material contractual obligations as of December 31, 2025 include: (i) future minimum lease payments under our San Diego, California office lease of $184,167 in aggregate through December 31, 2028, with $47,527 due in 2026; (ii) principal and accrued interest on related party notes payable aggregating $500,100 in principal, maturing May 19, 2026; and (iii) the PIPE convertible note with a principal amount of $10.9 million maturing December 8, 2026. We have also entered into a prepaid services agreement with our CRO, Prevail InfoWorks Inc., with a prepaid balance of approximately $2.0 million as of December 31, 2025, to be applied against future clinical trial costs.
Segment Information
Following the completion of the MindWave Merger on December 1, 2025, the Company operates in two segments: (i) the biopharmaceutical segment, which is focused on the clinical development of Apitox through Lokahi, and (ii) the digital asset operations segment, which encompasses the holding, sale, and advancement of the MindWaveDAO blockchain and related digital assets through MindWave.
Critical Accounting Estimates
The preparation of our consolidated financial statements requires management to make estimates and assumptions that affect reported amounts. We consider the following estimates to be the most critical to understanding our financial results and condition:
Fair Value of Digital Assets. We account for digital assets, including BTC, USDT, and NILA Tokens, at fair value in accordance with ASU 2023-08. BTC and USDT are classified within Level 1 of the fair value hierarchy based on quoted prices in active markets. NILA Tokens are classified within Level 2, as they trade on a limited number of centralized exchanges with modest daily trading volume. Changes in the fair value of digital assets are recognized in earnings each reporting period. Given the significant concentration of digital assets on our balance sheet of approximately $149.9 million as of December 31, 2025, changes in the fair value of these assets, particularly BTC and NILA Tokens, could have a material impact on our reported results.
45
Fair Value of Stock-Based Compensation. We measure the fair value of stock options using the Black-Scholes option-pricing model, which requires significant estimates including expected volatility, expected term, and risk-free interest rate. Changes in these assumptions could materially affect the amount of stock-based compensation expense recognized.
Fair Value of Derivative Liability. The derivative liability bifurcated from the PIPE convertible note is measured at fair value each reporting period. The variable conversion feature, which allows the noteholder to convert at a 20% discount to the minimum VWAP over a five-day lookback period, is sensitive to changes in our stock price and volatility. Changes in the fair value of the derivative are recognized in earnings and could be material in future periods.
Going Concern Assessment. Management’s assessment of the Company’s ability to continue as a going concern requires significant judgment regarding future capital raises, the availability of remaining PIPE tranches, and the expected pace of operating expenditures. Changes in these assumptions could affect the going concern conclusion and related disclosures.
Purchase Price Allocation. The allocation of consideration in the MindWave Merger required management to estimate the fair values of acquired digital assets and assumed liabilities at the acquisition date. The fair values of NILA Tokens, in particular, involved the use of Level 2 observable inputs and management judgment regarding market activity and trading volumes.
Recently Issued Accounting Pronouncements
We adopted ASU 2023-08, Accounting for and Disclosure of Crypto Assets, effective upon the acquisition of digital assets in connection with the MindWave Merger on December 1, 2025. Under ASU 2023-08, in-scope crypto assets that meet the definition of an intangible asset and are fungible are measured at fair value with changes recognized in earnings each period. The adoption of ASU 2023-08 did not have a cumulative effect on periods prior to adoption, as the Company had no digital asset holdings prior to the Merger.
Recent Developments
On April 24, 2026, the Company, MindWave, and Lokahi Therapeutics, Inc., a Nevada corporation (“Lokahi” and, together with the Company and MindWave, the “Company Parties”), together with Erik Emerson, individually and in his capacity as Bio Business Representative under the Merger Agreement, entered into a Confidential Settlement and Mutual Release Agreement (the “Settlement Agreement”) with Inscobee Inc., a South Korean corporation (“Inscobee”), and Apimeds Inc., a South Korean corporation and wholly owned subsidiary of Inscobee (“Apimeds Korea” and, together with Inscobee, the “Inscobee Parties”). Concurrently with the Settlement Agreement, the Company Parties and the Inscobee Parties also entered into a Side Letter Agreement (Merger Unwind Conditions) (the “Side Letter”), which is incorporated into and forms part of the Settlement Agreement.
The Settlement Agreement resolves all outstanding disputes among the parties arising from the Merger Agreement and related transactions.
Also on April 30, 2026, the Company entered into a Forbearance Agreement (the “Forbearance Agreement”) with Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B (the “Investor”), which holds a senior convertible note in the aggregate original principal amount of $11,000,000 (the “Existing Note”), issued pursuant to a Securities Purchase Agreement, dated December 1, 2025 (the “Securities Purchase Agreement”).
Pursuant to the Forbearance Agreement, the Investor has agreed to forbear from exercising any of its rights or remedies under the Existing Note with respect to certain existing events of default (collectively, the “Existing Defaults”) during the period commencing on the date of the Forbearance Agreement through and including June 30, 2026 (or such later date as the Investor may elect in its sole discretion) (the “Forbearance Period”).
The Settlement Agreement, the Side Letter, and the Forbearance Agreement are described in the Company’s Current Report on Form 8-K filed with the SEC on May 3, 2026, and are incorporated herein by reference.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
As a smaller reporting company, we are not required to provide the information required by this Item.
Item 8. Financial Statements and Supplementary Data
This information appears following Item 15 of this Report and is incorporated herein by reference.
46
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
(a) Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as such term is defined under Rule 13a-15(e) promulgated under the Exchange Act, designed to ensure that information required to be disclosed in our reports filed pursuant to the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and our principal financial officer, as appropriate to allow timely decisions regarding required disclosures.
In designing and evaluating the disclosure controls and procedures, we recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and we were required to apply our judgment in evaluating the cost-benefit relationship of possible controls and procedures. We have carried out an evaluation as of December 31, 2025 under the supervision, and with the participation, of our management, including our Chief Executive Officer, Dr. Vin Menon (who serves as our principal executive officer), and our Chief Financial Officer, Erick Frim (who serves as our principal financial officer), of the effectiveness of the design and operation of our disclosure controls and procedures.
Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were not effective as of December 31, 2025 in providing reasonable assurance of achieving the desired control objectives due to the material weaknesses in internal control over financial reporting described below.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting refers to the process designed by, or under the supervision of, our principal executive officer and principal financial officer, and effected by our Board, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
| (1) | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
| (2) | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and |
| (3) | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisitions, use or disposition of our assets that could have a material effect on the financial statements. |
Material Weakness in Internal Control Over Financial Reporting
Management has identified a material weakness in the Company’s internal control over financial reporting. Due to the Company’s limited number of accounting personnel, the Company does not maintain adequate segregation of duties in certain key processes. As a result, certain individuals have responsibility for multiple aspects of transactions, including authorization, recording, and review, which increases the risk that a material misstatement of the financial statements could occur and not be prevented or detected on a timely basis.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the Company’s financial statements will not be prevented or detected on a timely basis.
Management has concluded that this control deficiency constitutes a material weakness. Accordingly, management has determined that the Company’s internal control over financial reporting was not effective as of the date of this Annual Report.
As a smaller reporting company with limited resources, the Company expects that segregation of duties will remain a challenge; however, management is implementing compensating controls, including increased management review and oversight procedures, to mitigate the risks associated with this material weakness
47
Internal control over financial reporting has inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
We have conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2025, based on the framework established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO Framework”). This assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of those controls.
A material weakness in internal controls is a deficiency in internal control, or combination of control deficiencies, that adversely affects our ability to initiate, authorize, record, process, or report external financial data reliably in accordance with GAAP such that there is more than a remote likelihood that a material misstatement of our annual or interim financial statements that is more than inconsequential will not be prevented or detected. In the course of making our assessment of the effectiveness of internal controls over financial reporting, we identified material weaknesses in our internal control over financial reporting. Specifically, we do not have sufficiently documented procedures or control activities in place to support a reliable financial reporting process. This includes an absence of controls over the review and approval of journal entries, segregation of duties, reconciliations, and other fundamental accounting processes.
Based on our assessment under the criteria described above, we have concluded that our internal control over financial reporting was not effective as of December 31, 2025.
(b) Changes in Internal Control Over Financial Reporting
There were no changes in the Company’s internal controls over financial reporting that occurred during the year ended December 31, 2025 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting. The Company continues to review its disclosure controls and procedures, including its internal control over financial reporting, and may from time to time make changes aimed at enhancing their effectiveness and to ensure that the Company’s systems evolve with its business.
Item 9B. Other Information
Item 9C. Disclosure Regarding Foreign Jurisdiction that Prevents Inspections
Not applicable.
48
PART III - OTHER INFORMATION
Item 10. Directors, Executive Officers and Corporate Governance
Information Regarding Directors and Executive Officers
The following table sets forth information regarding the Company’s executive officers and non-employee directors.
| Name | Age | Position | ||
| Dr. Vin Menon | 48 | Chief Executive Officer | ||
| Erick Frim | 68 | Chief Financial Officer | ||
| Christopher Kim, MD. | 74 | Chief Medical Officer | ||
| Dr. Bennett Weintraub, PhD. | 56 | Director | ||
| Carol O’Donnell | 67 | Director | ||
| Elona Kogan | 55 | Director |
Dr. Vin Menon — Chief Executive Officer
Dr. Vin Menon has been our Chief Executive Officer since December 2025. Dr. Menon is a veteran in the technology services industry, who can be credited with the strategic direction behind several disruptive technology companies. In the corporate world, he has held various leadership positions at multinational corporations like HP & Compaq with global responsibilities. Driven by his passion for technology and innovation, Dr. Menon has been a forerunner in technological innovation and has helped create the business ecosystem of disruptive technologies and high-growth companies. His experience has helped him in the technology space as an entrepreneur and advisor, leading several startups from inception to meteoric growth across continents. Dr. Menon’s proven track record of setting up motivated and high caliber teams in the technology and services industry, establishing development centers from scratch to scale, and building company competencies led him to being awarded ‘Entrepreneur of the Year 2012’ by Rotary-ASME, ‘Outstanding Entrepreneur Award 2011’ by APEA, the ’Spirit of Enterprise 2010’ by SOE Singapore. Dr. Menon was also selected as a ‘Leading Indian Entrepreneur of the Year 2010’ by the Singapore Indian Chamber of Commerce. From the years 2021 to 2023 he co-founded and served as Strategic Advisor to CGCX, a Fintech, decentralized finance, and digital assets platform, where he provided strategic advisory services and growth initiatives. He currently serves as Chief Executive Officer of AQUAE Impact (AQUAE Impact Exchange Co. L.L.C/AQUAE Impact), a sustainable financial and environmental assets platform that uses blockchain technology and artificial intelligence, which he co-founded and currently forms part of its executive leadership providing oversight of product and sustainability initiatives. He also currently serves as Chief Executive Officer of AQUAE Labs Pte Ltd, which is the research and development and technology arm of AQUAE Impact, where he provides product and technology leadership, measurement, reporting, and verification of environmental credits. Additionally, he currently occupies the role of Strategic Advisor of TechyTrade FZ-LLC, which is a bitcoin-backed company that operates in the digital asset and treasury innovation space. Dr. Menon is also a champion of techno-preneurship and was serving on the Board of Directors of the Spirit of Enterprise (SOE) and the Mentoring Programme under Action Community for Entrepreneurship (ACE) by SPRING Singapore. Moreover, he completed his bachelor’s degree in computer applications from India, with first-class honors. He has also completed the following programs: Advanced Management Program (AMP) at NTU-Berkeley (Haas Business School, California) and Advanced Management Program (AMP) at The Wharton School (University of Pennsylvania, USA) specialized in Finance. Dr. Menon obtained an EMBA from the Nanyang Technological University (NTU) in Singapore. Lastly, he completed his PhD, Blockchain for Impact in Healthcare from The Open International University for Complementary Medicine in collaboration with Al-Farabi Kazakh National University, Kazakhstan 2019. We believe that these experiences provide Dr. Menon with the skills necessary to lead the Company as its Chief Executive officer, including overseeing the Company’s strategy, operations, financial performance, and overall corporate governance.
49
Erick Frim — Chief Financial Officer
Erick Frim has over 40 years of experience as an accountant, financial executive and consultant. Mr. Frim joined Apimeds as CFO in July of 2025. In 2019, he joined CFO Squad and as a partner in the CFO Squad, LLC Mr. Frim advised clients on technical accounting and regulatory compliance, assisting numerous companies with their initial public offerings. Prior to the CFO Squad, Mr. Frim served as a director in the public company audit practice of EisnerAmper LLP. Mr. Frim as also served as a financial executive for digital media pioneer DIVA Systems Corporation. A former CPA, he has a BS in Accounting from Ball State University.
Christopher Kim, MD. — Chief Medical Officer
Dr. Christopher Kim has been our Chairman and Chief Medical Officer since our inception and served as our interim Chief Executive Officer from July 2022 to September 2023. Dr. Kim is the inventor and developer of Apitox and the founder of Apimeds Korea, where he has served as a director since its inception. Mr. Kim served as the Chief Executive Officer of Apimeds Korea from May 2003 to August 2011. Prior to founding Apimeds Korea, Dr. Kim lead with the support of Guju Pharmaceuticals, clinical trials for Apitoxin in Korea, which was approved by the Korea Food and Drug Administration in 2003 for relief of pain and inflammation for patients with Osteoarthritis. In 2005, he began focusing on the clinical development of Apitox in the United States, including the first of two-Phase III clinical studies for Osteoarthritis. Prior to his time with Apimeds Korea, Dr. Kim served as the President of the International Pain Institute of New Jersey from January 1983 to May 2003, a center for chronic pain and other disabling diseases that conducted clinical research and provided treatment. He served as a professor at Biomedical Center, CHA Graduate School of Medicine in Korea from March 2005 to February 2017. Dr. Kim is a licensed physician in New Jersey, New York and Korea and a Pain Medicine Specialist (American Board). Over the past twenty years, Dr. Kim has treated thousands of chronically disabled patients with autoimmune diseases, including MS. Dr. Kim received his medical degree from the School of Medicine, CN University in Korea.
We believe Dr. Kim’s extensive experience in pharmaceutical development and the biopharmaceutical industry, as well as his research and treatment of autoimmune diseases, and institutional knowledge of our product candidate, qualifies him to serve as of Chief Medical Officer.
Independent Directors:
Dr. Bennett Weintraub, PhD.
Dr. Weintraub has served as a director since October 2024. Dr. Weintraub currently serves as the President of inThought Research (“inThought”), a healthcare business intelligence consulting firm which he founded in 2009. inThought provides business development support, competitive intelligence monitoring, medical conference coverage, and other services both to professional investors and to pharma/biotech companies. Dr. Weintraub has also served as the Chief Scientific Officer of inPhronesis since 2018.
After completing his training in immunology and biochemistry, Dr. Weintraub co-founded Biotech Tracker, an online tool for investors, where he served as a financial analyst from 2000 to 2008. From 2006 to 2008, Dr. Weintraub served as an analyst at Reuters Insight, providing analysis of drug development and trends in medicine to professional investors. Dr. Weintraub served as a licensed security analyst with Variant Research from 2005 to 2006.
From 1999 to 2000, Dr. Weintraub was senior scientific editor for the biology research journals Cell and Molecular Cell. Dr. Weintraub performed biochemistry and immunology research at Stanford University and at the John Curtin School of Medical Research in Canberra, Australia. He earned his doctorate in Biology from the University of California, San Diego, and a Bachelor of Science in Life Science from the Massachusetts Institute of Technology.
We believe Mr. Weintraub’s extensive science background qualifies him to serve on our Board of Directors.
From 1999 to 2000, Dr. Weintraub was senior scientific editor for the biology research journals Cell and Molecular Cell. Dr. Weintraub performed biochemistry and immunology research at Stanford University and at the John Curtin School of Medical Research in Canberra, Australia. He earned his doctorate in Biology from the University of California, San Diego, and a Bachelor of Science in Life Science from the Massachusetts Institute of Technology.
We believe Mr. Weintraub’s extensive science background qualifies him to serve on our Board.
50
Carol O’Donnell
Carol O’Donnell has served as a director since October 2024. Ms. O’Donnell is currently a Director and Member of the Audit Committee of Sono-Tek Corporation (NASDAQ: SOTK), where she has served since November 2018. Prior to that, she served as General Counsel to Boothbay Fund Management LLC, a registered investment adviser, from December 2019 through May 2021. Ms. O’Donnell joined Protégé Partners and MOV37, an industry leading firm investing in and seeding smaller and emerging hedge fund managers in April 2016 and has served as Chief Executive Officer since January 2018. Prior to joining Protégé Partners and MOV37, Ms. O’Donnell was the Director of Legal and Compliance with DARA Capital US, Inc., a Swiss-owned boutique registered investment advisory and wealth management firm from January 2013 to March 2016. She served as General Counsel and Chief Compliance Officer of each of the Permal Group and Framework Investment Group from June 2004 through February 2011 and from January 2002 to May 2004, respectively. She also served as a director of FSI Low Beta from 2012 to 2021. Ms. O’Donnell was named one of the Top 50 Women in Hedge Funds in September 2018 and is currently admitted to practice law in the State of Connecticut.
We believe Ms. O’Donnell’s extensive experience in the financial industry qualifies her to serve on our Board of Directors.
Elona Kogan
Elona Kogan has served as a director since October 2024. Beginning in August 2024, Most recently, Ms. Kogan served as the Chief Legal Officer of Terns Pharmaceutical, Inc. (Nasdaq: TERNS), a publicly traded biopharmaceutical company, Prior to joining us, from November 2020 through August 2024, Ms. Kogan served as the General Counsel and Chief Legal Officer of Seer Inc. (Nasdaq: SEER), a publicly traded life science company. From May 2018 through August 2021, Ms. Kogan served as a director of Cardax, Inc., a biotechnology company operating in the wellness health care space. From March 2019 through August 2020, Ms. Kogan served as the General Counsel of Selecta Biosciences, Inc., a clinical-stage biotechnology company. Ms. Kogan is a graduate of Southwestern University School of Law. Ms. Kogan graduated from Columbia University, Barnard College, with a B.A. in Economics.
We believe Ms. Kogan’s extensive experience as an operating executive in biopharmaceutical and life science space, in addition to her experience leading government relations, and serving as general counsel and chief legal officer of other publicly traded companies qualifies her to serve on our Board of Directors.
51
Family Relationships
There are no family relationships among any of our executive officers or directors.
Involvement in Certain Legal Proceedings
To the best of our knowledge, none of our executive officers or directors were involved in any legal proceedings described in Item 401(f) of Regulation S-K in the past ten years.
Compliance with Section 16(a) of the Exchange Act
Section 16(a) of the Securities Exchange Act of 1934, requires our directors, executive officers and persons who own more than 10% of our common stock to file with the SEC initial reports of ownership and reports of changes in ownership of common stock and other of our equity securities. During the year ended December 31, 2025, our officers, directors and 10% stockholders were not required to make filings pursuant to Section 16(a).
Code of Business Conduct and Ethics
In accordance with the information required by this Item 10 relating to the code of ethics required by Item 406 of Regulation S-K, the Company has a Code of Business Conduct and Ethics (the “Code”), which applies to its directors, officers (including its principal executive officer, the principal financial officer and principal accounting officer), and all other employees (collectively, the “Covered Persons” and each a “Covered Person”). The full text of the Code is available on the “Investors” section of the Company’s website. The Company intends to satisfy the SEC’s requirements regarding amendments to, or waivers from, the Code by posting such information on its website or by filing a Current Report on Form 8-K to disclose such information.
Procedures for Stockholders to Recommend Director Nominees
The Company’s Amended and Restated Bylaws (the “Bylaws”) were originally adopted on May 12, 2020, amended and restated on April 11, 2025, and further amended on October 15, 2025. The Bylaws authorize the Board of Directors to designate one or more committees, each consisting of one or more directors. On February 7, 2025, the Board established the Nominating and Corporate Governance Committee and adopted a written charter for such committee. Pursuant to the Nominating and Corporate Governance Committee’s charter, the committee may, if it deems appropriate, establish procedures to be followed by stockholders in submitting recommendations for Board candidates.
Committees of the Board of Directors
Our Board of Directors established three standing committees: an audit committee, a compensation committee and the nominating and corporate governance committee.
52
Audit Committee
The members of the audit committee are Carol O’Donnell (chair), Elona Kogan, and Dr. Bennett Weintraub. Upon his appointment to the Board of Directors, Amir Dossal is expected to be appointed as a member of the audit committee. Our Board of Directors has determined that each of the members of the audit committee is an “independent director” as defined by and meet the other requirements of the NYSE American listing standards and applicable SEC rules.
Each member of the audit committee is financially literate. Carol O’Donnell qualifies as an “audit committee financial expert” as defined in applicable SEC rules and meets the financial sophistication requirements of the NYSE rules. In making this determination, our Board of Directors considered Ms. O’Donnell’s previous and current experience in actively supervising individuals in financial and accounting roles. The primary purpose of the audit committee is to discharge the responsibilities of the Board of Directors with respect to our accounting, financial, and other reporting and internal control practices and to oversee our independent registered accounting firm. Specific responsibilities of our audit committee include:
| ● | assisting board oversight of (1) the integrity of our financial statements, (2) our compliance with legal and regulatory requirements, (3) our independent auditor’s qualifications and independence, and (4) the performance of our internal audit function and independent auditors; |
| ● | reviewing the appointment, compensation, retention, replacement, and oversight of the work of the independent auditors and any other independent registered public accounting firm engaged by us; |
| ● | pre-approving all audit and non-audit services to be provided by the independent auditors or any other registered public accounting firm engaged by us, and establishing pre-approval policies and procedures; |
| ● | reviewing and discussing with the independent auditors all relationships the auditors have with us in order to evaluate their continued independence; |
| ● | setting clear hiring policies for employees or former employees of the independent auditors; |
| ● | setting clear policies for audit partner rotation in compliance with applicable laws and regulations; |
| ● | obtaining and reviewing a report, at least annually, from the independent auditors describing (1) the independent auditor’s internal quality-control procedures and (2) any material issues raised by the most recent internal quality-control review, or peer review, of the audit firm, or by any inquiry or investigation by governmental or professional authorities, within the preceding five years respecting one or more independent audits carried out by the firm and any steps taken to deal with such issues; |
| ● | meeting to review and discuss our annual audited financial statements and quarterly financial statements with management and the independent auditor, including reviewing our specific disclosures under “Management’s Discussion and Analysis of Financial Condition and Results of Operations”; |
| ● | reviewing and approving any related party transaction required to be disclosed pursuant to Item 404 of Regulation S-K promulgated by the SEC prior to us entering into such transaction; and |
| ● | reviewing with management, the independent auditors, and our legal advisors, as appropriate, any legal, regulatory or compliance matters, including any correspondence with regulators or government agencies and any employee complaints or published reports that raise material issues regarding our financial statements or accounting policies and any significant changes in accounting standards or rules promulgated by the Financial Accounting Standards Board, the SEC or other regulatory authorities. |
Compensation Committee
The members of the compensation committee are Carol O’Donnell (chair) and Dr. Bennett Weintraub. Upon his appointment to the Board of Directors, Amir Dossal is expected to be appointed as a member of the compensation committee. Under the NYSE American listing standards and applicable SEC rules, we are required to have at least two members of the compensation committee, all of whom must be independent. Our Board of Directors has determined that each of the members of the compensation committee is an “independent director” as defined by and meet the other requirements of the NYSE American listing standards and applicable SEC rules.
53
Specific responsibilities of our compensation committee include:
| ● | reviewing and approving on an annual basis the corporate goals and objectives relevant to our Chief Executive Officer’s compensation, evaluating our Chief Executive Officer’s performance in light of such goals and objectives and determining and approving the remuneration (if any) of our Chief Executive Officer based on such evaluation; |
| ● | reviewing and making recommendations to our Board of Directors with respect to (or approving, if such authority is so delegated by our board of directors) the compensation, and any incentive-compensation and equity-based plans that are subject to board approval of all of our other officers; |
| ● | reviewing our executive compensation policies and plans; |
| ● | implementing and administering our incentive compensation equity-based remuneration plans; |
| ● | assisting management in complying with our proxy statement and annual report disclosure requirements; |
| ● | approving all special perquisites, special cash payments and other special compensation and benefit arrangements for our officers and employees; |
| ● | producing a report on executive compensation to be included in our annual proxy statement; and |
| ● | reviewing, evaluating and recommending changes, if appropriate, to the remuneration for directors. |
Nominating and Governance Committee
The members of the nominating and corporate governance committee are Elona Kogan (chair) and Carol O’Donnell. Upon his appointment to the Board of Directors, Amir Dossal is expected to be appointed as a member of the nominating and corporate governance committee. Our Board of Directors has determined that each of the members of the nominating and corporate governance committee is an “independent director” as defined by and meet the other requirements of the NYSE American listing standards and applicable SEC rules.
Specific responsibilities of our nominating and corporate governance committee include:
| ● | identifying and evaluating candidates, including the nomination of incumbent directors for reelection and nominees recommended by stockholders, to serve on our Board of Directors; |
| ● | considering and making recommendations to our Board of Directors regarding the composition and chairmanship of the committees of our Board of Directors; |
| ● | developing and making recommendations to our Board of Directors regarding corporate governance guidelines and matters; and |
| ● | overseeing periodic evaluations of the board of directors’ performance, including committees of the Board of Directors. |
In general, in identifying and evaluating nominees for director, the committee considers educational background, diversity of professional experience, knowledge of our business, integrity, professional reputation, independence, wisdom, and the ability to represent the best interests of our stockholders
Insider Trading Policy
The Company has an insider trading policy (the “Insider Trading Policy”) which prohibits Covered Persons from buying or selling the Company’s securities while the Covered Person is aware of material nonpublic information about the Company. The Company believes that its Insider Trading Policy is reasonably designed to promote compliance with insider trading laws, rules and regulations, and any applicable listing standards. A copy of the Insider Trading Policy is filed as Exhibit 19.1 to this Annual Report.
54
Item 11. Executive Compensation
The following discussion contains forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs. The actual amount and form of compensation and the compensation policies and practices that we adopt in the future may differ materially from currently planned programs as summarized in this discussion.
As an “emerging growth company,” we have opted to comply with the executive compensation disclosure rules applicable to “smaller reporting companies,” as such term is defined in the rules promulgated under the Securities Act. Accordingly, we are required to provide a Summary Compensation Table, as well as limited narrative disclosures regarding executive compensation for our last two completed fiscal years and an Outstanding Equity Awards at Fiscal Year End Table for our last completed fiscal year. These reporting obligations extend only to “named executive officers.” Individuals we refer to as our “named executive officers” include (i) all individuals serving as our principal executive officer during the fiscal year ended December 31, 2025 and (ii) our two most highly compensated executive officers, as defined in Exchange Act Rule 3b-7, other than our principal executive officer, who were serving as executive officers at the end of the fiscal year ended December 31, 2025, whose salary and bonus for services rendered in all capacities exceeded $100,000 during the fiscal year ended December 31, 2025.
The Company’s “named executive officers” consist of (i) the Company’s Chief Executive Officer, (ii) the Company’s Chief Financial Officer, and (iii) each other executive officer who served during fiscal year 2025 and whose compensation exceeded $100,000, if any.
Summary Compensation Table
The following table sets forth information concerning compensation earned by each of the Company’s named executive officers for the fiscal years ended December 31, 2025 and 2024:
| * | All option awards derived using Black-Scholes Pricing Model |
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Option Awards ($) | Nonequity Incentive Plan Compensation ($) | All Other Compensation ($) | Total ($) | ||||||||||||||||||||
| Erik Emerson | 2025 | $ | 250,000 | — | $ | 699,703.49 | — | $ 1,275,000 | (2) | $ | 2,224,703.49 | ||||||||||||||||
| Former Chief Executive Officer | 2024 | 300,000 | — | — | — | — | 300,000 | ||||||||||||||||||||
| Erick Frim | 2025 | $ | 10,128 | (1) | — | — | — | — | $ | 10,128 | |||||||||||||||||
| Chief Financial Officer (Advisory) | 2024 | — | — | — | — | — | — | ||||||||||||||||||||
| Christopher Kim, MD | 2025 | $ | 200,000 | — | $ | 4,045.80 | — | $ 425,000 | (2) | $ | 629,045.80 | ||||||||||||||||
| Chief Medical Officer | 2024 | 96,000 | — | — | — | — | 96,000 | ||||||||||||||||||||
| (1) | This figure represents compensation paid for six months, reflecting that the executive’s compensation commenced during fiscal year 2025. |
| (2) | These figures represent common stock issued to executives and valued at the observable market price at closing of the grant date: May 16, 2025. |
Narrative to Summary Compensation Table
Our executive compensation program is based on a pay for performance philosophy. Compensation for our executive officers is composed primarily of the following main components: base salary, bonus, and equity incentives in the form of stock options. Like all full-time employees, our executive officers are eligible to participate in our health and welfare benefit plans.
55
Employment Agreement with Erik Emerson
The Company entered into an employment agreement with Erik Emerson on September 21, 2023 (the “Emerson Employment Agreement”). Pursuant to the Emerson Employment Agreement, he would serve as the Company’s Chief Executive Officer and receive a yearly salary of $300,000. Mr. Emerson’s employment continued for one year from the date of execution and shall automatically renew for successive one year periods in the event of a closing on a public offering of the company during the initial term unless either party gives 30 days’ written notice of its intent not to renew the Emerson Agreement prior to the end of the then-current term.
Mr. Emerson may be terminated with or without Cause (as defined in the Emerson Agreement) upon thirty (30) days’ written notice to Mr. Emerson.
On November 13, 2025, the Company entered into an amendment to the Emerson Employment Agreement, increasing Mr. Emerson’s annual base salary to $500,000. It also provided that if Mr. Emerson is terminated by the Company without cause, Mr. Emerson will be entitled to severance payment equal to twenty-four (24) months of base salary and benefits and immediate vesting of all unvested equity, subject to Mr. Emerson’s execution of a release of claims against the Company. The amendment further clarifies that, upon termination by the Company for cause or by Mr. Emerson without good reason, any unvested equity will be automatically forfeited without payment or consideration by the Company.
In connection with and pursuant to the Merger Agreement, Mr. Emerson resigned from his position as Chief Executive Officer effective as of December 1, 2025.
Stock Option Award to Dr. Christopher Kim
On May 12, 2020 the Company granted Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer, a non-qualified stock option award to purchase 138,900 shares of the Company’s common stock at an exercise price of $11.28 per share. The option vested in three equal installments and vested fully on May 12, 2023. The options have a term of ten years from the date of grant and shall terminate at the expiration of that period, unless it is terminated at an earlier date pursuant to the provisions of the option grant agreement between the Company and Dr. Kim.
Outstanding Equity Awards at Fiscal-Year End 2025
There were no outstanding equity-based awards of the Company held by the named executive officer as of December 31, 2025.
Policies and Practices for Granting Certain Equity Awards
We do not schedule equity award grants in anticipation of the release of material nonpublic information, nor do we time the release of material nonpublic information based on equity grant dates.
Director Compensation Table
| * | All option awards derived using Black-Scholes Pricing Model |
| Name & Principal Position | Year | Nonemployee Compensation (Director Fees) | Option Awards ($) | Total | ||||||||||
| Carol O’Donnell | 2025 | $ | 16,000 | $ | 29,717.63 | $ | 45,717.63 | |||||||
| Director | ||||||||||||||
| Hankil Yoon | 2025 | $ | 10,000 | $ | 29,717.63 | $ | 39,717.63 | |||||||
| Director | ||||||||||||||
| Elona Kogan | 2025 | $ | 13,000 | $ | 29,717.63 | $ | 42,717.63 | |||||||
| Director | ||||||||||||||
| Bennett Weintraub | 2025 | $ | 10,000 | $ | 29,717.63 | $ | 39,717.63 | |||||||
| Director | ||||||||||||||
56
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters Securities
Authorized for Issuance under Share-Based Compensation Plans
Equity Compensation Plan Information
The following table sets forth, as of December 31, 2025, information regarding awards previously granted and outstanding, and securities authorized for future issuance, under the Company’s equity compensation plans.
| Plan Category | Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants or Rights | Weighted-Average Exercise Price of Outstanding Options, Warrants or Rights | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Outstanding Options, Warrants, or Rights) | |||||||||
| Equity compensation plans approved by shareholders | — | — | 1,000,000 | |||||||||
| Equity compensation plans not approved by shareholders | - | |||||||||||
Apimeds Pharmaceuticals US, Inc. 2024 Equity Incentive Plan
On September 18, 2024, we adopted an equity incentive plan for our employees, the Apimeds Pharmaceuticals US, Inc. 2024 Equity Incentive Plan (the “2024 Equity Incentive Plan”). The purposes of the Equity Incentive Plan are to provide additional incentives to selected employees, directors and independent contractors of, and consultants to, the Company or its affiliates, to strengthen their commitment, motivate them to faithfully and diligently perform their responsibilities and to attract and retain competent and dedicated persons who are essential to the success of our business and whose efforts will impact our long-term growth and profitability.
On December 1, 2025, in connection with the Merger, the Company’s stockholders approved an amendment to the 2024 Plan to increase the aggregate number of shares of common stock authorized for issuance under the 2024 Plan from 1,538,462 shares to 2,096,679 shares (the “2024 Plan Share Increase”). The 2024 Plan Share Increase was necessary as the 2024 Plan did not have a sufficient number of authorized shares to be issued in connection with the transactions contemplated by the Merger Agreement.
Awards
The 2024 Equity Incentive Plan allows the Company to make equity and equity-based incentive awards to officers, employees, directors, consultants, and advisors. The Board anticipates that providing such persons with a direct stake in the Company will assure a closer alignment of the interests of such individuals with those of the Company and its stockholders, thereby stimulating their efforts on the Company’s behalf and strengthening their desire to remain with the Company.
The 2024 Equity Incentive Plan provides for the grant of non-qualified stock options, incentive stock options, stock appreciation rights, restricted stock, restricted stock units, stock bonus awards, and performance compensation awards. All awards will be set forth in an award agreement which will detail all terms and conditions of the awards, including any applicable vesting and payment terms and post-termination exercise limitations.
57
A brief description of each award type follows.
| ● | Non-Qualified Stock Options means the right to purchase shares pursuant to terms and conditions that are not intended to be, or do not qualify as, an Incentive Stock Options; |
| ● | Incentive Stock Options means the right to purchase shares pursuant terms and conditions that are intended to qualify as, and that satisfy the requirements applicable to, an incentive equity option within the meaning of Code Section 422 of the United States Internal Revenue Code of 1986, as amended; |
| ● | Stock Appreciation Rights means a right, designated as an SAR, to receive the appreciation in the fair market value of shares; | |
| ● | Restricted Stock means an award of shares subject to vesting conditions; |
| ● | Restricted Stock Units shall mean a right to receive shares or cash upon vesting; |
| ● | Stock Bonus Awards means unrestricted common stock, or other awards denominated in common stock, either alone or in tandem with other awards; and |
| ● | Performance Compensation Awards means an award granted to a participant that entitles the participant to delivery of shares or cash upon achievement of performance goals. |
1,000,000 shares of common stock have initially been reserved for the issuance of awards under the 2024 Equity Incentive Plan (the “Initial Limit”). The Initial Limit is subject to adjustment in the event of a reorganization, recapitalization, reclassification, stock split, stock dividend, reverse stock split or other similar change in the Company’s capitalization. The maximum aggregate number of shares of common stock of the Company that may be issued upon exercise of incentive stock options under the 2024 Equity Incentive Plan shall not exceed the Initial Limit, as adjusted. Shares underlying any awards under the 2024 Equity Incentive Plan that are forfeited, cancelled, held back upon exercise of an option or settlement of an award to cover the exercise price or tax withholding, satisfied without the issuance of stock or otherwise terminated (other than by exercise) will be added back to the shares available for issuance under the 2024 Equity Incentive Plan and, to the extent permitted under Section 422 of the Code and the regulations promulgated thereunder, the shares that may be issued as incentive stock options.
The 2024 Equity Incentive Plan is currently administered by a committee of at least two people as the Board may appoint to administer the 2024 Equity Incentive Plan or, if no such committee has been appointed by the Board, the Board, pursuant to the terms of the 2024 Equity Incentive Plan (the “Committee”). The plan administrator, which initially will be the Committee, has full power to select, from among the individuals eligible for awards, the individuals to whom awards will be granted, to make any combination of awards to participants, and to determine the specific terms and conditions of each award, subject to the provisions of the 2024 Equity Incentive Plan. The plan administrator may delegate to a committee consisting of one or more officers of the Company, the authority to awards to individuals who are not subject to the reporting and other provisions of Section 16 of the Exchange Act and not members of the delegated committee, subject to certain limitations and guidelines.
Persons eligible to participate in the 2024 Equity Incentive Plan will be officers, employees, non-employee directors, consultants, and advisors of the Company and its subsidiaries as selected from time to time by the plan administrator in its discretion. As of the date of this Annual Report, approximately 12 individuals are eligible to participate in the 2024 Equity Incentive Plan, which includes approximately two officers, no employees who are not officers, five non-employee directors, and five consultants/independent contractors.
58
Options
The 2024 Equity Incentive Plan permits the granting of both options to purchase common stock of the Company intended to qualify as incentive stock options under Section 422 of the Code and options that do not so qualify. Options granted under the 2024 Equity Incentive Plan will be non-qualified options if they fail to qualify as incentive stock options or exceed the annual limit on incentive stock options. Incentive stock options may only be granted to employees of the Company and its subsidiaries. Non-qualified options may be granted to any persons eligible to receive awards under the 2024 Equity Incentive Plan. The option exercise price of each option will be determined by the plan administrator but generally may not be less than 100% of the fair market value of the common stock of the Company on the date of grant or, in the case of an incentive stock option granted to a ten percent stockholder, 110% of such share’s fair market value. The term of each option will be fixed by the plan administrator and may not exceed ten years from the date of grant. The plan administrator will determine at what time or times each option may be exercised, including the ability to accelerate the vesting of such options.
Upon exercise of options, the option exercise price must be paid in full either in cash, by certified or bank check or other instrument acceptable to the plan administrator or by delivery (or attestation to the ownership) of shares of common stock of the Company that are beneficially owned by the optionee free of restrictions or were purchased in the open market. Subject to applicable law, the exercise price may also be delivered by a broker pursuant to irrevocable instructions to the broker from the optionee. In addition, the plan administrator may permit non-qualified options to be exercised using a “net exercise” arrangement that reduces the number of shares issued to the optionee by the largest whole number of shares with fair market value that does not exceed the aggregate exercise price.
Stock Appreciation Rights
The plan administrator may award stock appreciation rights subject to such conditions and restrictions as it may determine. Stock appreciation rights entitle the recipient to shares of common stock of the Company, or cash, equal to the value of the appreciation in the Company’s stock price over the exercise price. The exercise price generally may not be less than 100% of the fair market value of common stock of the Company on the date of grant. The term of each stock appreciation right will be fixed by the plan administrator and may not exceed ten years from the date of grant. The plan administrator will determine at what time or times each stock appreciation right may be exercised, including the ability to accelerate the vesting of such stock appreciation rights.
Restricted Stock and Restricted Stock Units
The plan administrator may award restricted shares of common stock of the Company and restricted stock units to participants subject to such conditions and restrictions as it may determine. These conditions and restrictions may include the achievement of certain performance goals and/or continued employment with the Company through a specified vesting period. The plan administrator may also grant shares of common stock of the Company that are free from any restrictions under the 2024 Equity Incentive Plan. Unrestricted stock may be granted to participants in recognition of past services or for other valid consideration and may be issued in lieu of cash compensation due to such participant. The plan administrator may grant dividend equivalent rights to participants that entitle the recipient to receive credits for dividends that would be paid if the recipient had held a specified number of shares of common stock of the Company.
Stock Bonus Awards
The plan administration may issue unrestricted common stock, or other awards denominated in common stock, under the 2024 Equity Incentive Plan to participants, either alone or in tandem with other awards, in such amounts as the plan administration shall from time to time in its sole discretion determine.
Performance Compensation Awards
The plan administrator may grant awards under the 2024 Equity Incentive Plan to participants, which may be cash-based, subject to the achievement of certain performance goals, including continued employment with the Company.
Other Material Features
The 2024 Equity Incentive Plan requires the plan administrator to make appropriate adjustments to the number of shares of common stock that are subject to the 2024 Equity Incentive Plan, to certain limits in the 2024 Equity Incentive Plan, and to any outstanding awards to reflect stock dividends, stock splits, extraordinary cash dividends and similar events.
59
Except as set forth in a stock award agreement issued under the 2024 Equity Incentive Plan, in the event of (i) a transfer of all or substantially all of the Company’s assets, (ii) a merger, consolidation or other capital reorganization or business combination transaction of the Company with or into another corporation, entity or person, or (iii) the consummation of a transaction, or series of related transactions, in which any person becomes the beneficial owner directly or indirectly, of more than 50% of Company’s then outstanding capital stock, each outstanding stock award (vested or unvested) will be treated as the plan administrator determines, which may include (a) Company’s continuation of such outstanding stock awards (if Company is the surviving corporation); (b) the assumption of such outstanding stock awards by the surviving corporation or its parent; (c) the substitution by the surviving corporation or its parent of new stock options or other equity awards for such stock awards; (d) the cancellation of such stock awards in exchange for a payment to the participants equal to the excess of (1) the fair market value of the shares subject to such stock awards as of the closing date of such corporate transaction over (2) the exercise price or purchase price paid or to be paid (if any) for the shares subject to the stock awards (which payment may be subject to the same conditions that apply to the consideration that will be paid to holders of shares in connection with the transaction, subject to applicable law); or (e) the opportunity for participants to exercise the stock options prior to the occurrence of the corporate transaction and the termination (for no consideration) upon the consummation of such corporate transaction of any stock options not exercised prior thereto.
The 2024 Equity Incentive Plan provides that a stock award may be subject to additional acceleration of vesting and exercisability upon or after a “Change in Control” (as defined in the 2024 Equity Incentive Plan) as may be provided in the award agreement for such stock award or as may be provided in any other written agreement between the Company or any affiliate and the participant, but in the absence of such provision, no such acceleration will occur.
Participants in the 2024 Equity Incentive Plan are responsible for the payment of any federal, state or local taxes that the Company or its subsidiaries are required by law to withhold upon the exercise of options or stock appreciation rights or vesting of other awards. The plan administrator may cause any tax withholding obligation of the Company or its subsidiaries to be satisfied, in whole or in part, by the applicable entity withholding from shares of common stock of the Company to be issued pursuant to an award shares with an aggregate fair market value that would satisfy the withholding amount due. The plan administrator may also require any tax withholding obligation of the Company or its subsidiaries to be satisfied, in whole or in part, by an arrangement whereby a certain number of shares issued pursuant to any award are immediately sold and proceeds from such sale are remitted to the Company or its subsidiaries in an amount that would satisfy the withholding amount due.
The 2024 Equity Incentive Plan generally does not allow for the transfer or assignment of awards, other than by will or by the laws of descent and distribution or pursuant to a domestic relations order; however, the plan administrator may permit the transfer of non-qualified stock options by gift to an immediate family member, to trusts for the benefit of family members, or to partnerships in which such family members are the only partners.
The plan administrator may amend or discontinue the 2024 Equity Incentive Plan and the plan administrator may amend or cancel outstanding awards for purposes of satisfying changes in law or any other lawful purpose, but no such action may materially and adversely affect rights under an award without the holder’s consent. Certain amendments to the 2024 Equity Incentive Plan will require the approval of the Company’s stockholders. Generally, without shareholder approval, (i) no amendment or modification of the 2024 Equity Incentive Plan may reduce the exercise price of any stock option or the strike price of any stock appreciation right, (ii) the plan administrator may not cancel any outstanding stock option or stock appreciation right where the fair market value of the common stock underlying such stock option or stock appreciation right is less than its exercise price and replace it with a new option or stock appreciation right, another award or cash and (iii) the plan administrator may not take any other action that is considered a “repricing” for purposes of the shareholder approval rules of the applicable securities exchange.
All awards granted under the 2024 Equity Incentive Plan will be subject to recoupment in accordance with any clawback policy that Company is required to adopt pursuant to the listing standards of any national securities exchange or association on which Company securities are listed or as is otherwise required by the U.S. Dodd-Frank Wall Street Reform and Consumer Protection Act or other applicable law. In addition, the Board may impose such other clawback, recovery or recoupment provisions in a stock award agreement as the Board determines necessary or appropriate.
No options or stock appreciation rights may be granted under the 2024 Equity Incentive Plan after the date that is ten years from the 2024 Equity Incentive Plan Effective Date. No awards under the 2024 Equity Incentive Plan have been made prior to the date of this Annual Report.
60
2025 Equity Incentive Plan
Apimeds Pharmaceuticals US, Inc. 2025 Equity Incentive Plan
On December 1, 2025, in connection with the Merger, the Company adopted the Apimeds Pharmaceuticals US, Inc. 2025 Equity Incentive Plan (the “2025 Plan”). The purpose of the 2025 Plan is to advance the interests of the Company, its subsidiaries, affiliates, and stockholders by providing an incentive to attract and retain the best qualified personnel to perform services for the Company, by motivating such individuals to contribute to the growth and the profitability of the Company by aligning such individuals with the interests of the Company’s stockholders, and by rewarding such individuals for their services by tying a significant portion of their compensation to the success of the Company.
Awards
The 2025 Plan allows the Company to make equity and equity-based incentive awards to officers, employees, directors, consultants, and advisors. The Board of Directors anticipates that providing such persons with a direct stake in the Company will assure a closer alignment of the interests of such individuals with those of the Company and its stockholders, thereby stimulating their efforts on the Company’s behalf and strengthening their desire to remain with the Company.
The 2025 Plan provides for the grant of stock options, stock appreciation rights, restricted stock, restricted stock units, and other stock-based awards. All awards are set forth in an award agreement which details all terms and conditions of the awards, including any applicable vesting and payment terms and post-termination exercise limitations.
A brief description of each award type follows:
| ● | Stock Options means an option granted under the 2025 Plan to purchase shares of common stock, whether designated as an Incentive Stock Option or a Nonqualified Stock Option; |
| ● | Stock Appreciation Rights means a right, designated as a SAR, to receive the appreciation in the fair market value of shares; |
| ● | Restricted Stock means shares, subject to a period of restriction or certain other specified restrictions (including, without limitation, a requirement that the participant remain continuously employed or provide continuous service for a specific period of time), granted under the 2025 Plan or issued pursuant to the early exercise of a Stock Option; |
| ● | Restricted Stock Units means an unfunded and unsecured promise to deliver shares, cash, other securities, or other property, subject to certain restrictions (including, without limitation, a requirement that the participant remain continuously employed or provide continuous service for a specific period of time) granted under the 2025 Plan; and |
| ● | Other Stock-Based Awards means any other awards not specifically described in the 2025 Plan that are valued in whole or in part by reference to, or are otherwise based on, shares and are created by the plan administrator pursuant to the 2025 Plan. |
The maximum aggregate number of shares that may be issued under the 2025 Plan is ten percent (10%) of the shares outstanding on December 1, 2025 (the “Plan Share Limit”). The shares subject to the 2025 Plan may be authorized, but unissued, or reacquired shares. On the first day of each calendar year during the term of the 2025 Plan, commencing on January 1, 2026 and continuing until (and including) January 1, 2035, the number of shares available under the Plan Share Limit automatically increases by a number equal to the lesser of (i) three percent (3%) of the total number of shares issued and outstanding on December 31 of the calendar year immediately preceding the date of such increase and (ii) a number of shares determined by the Board of Directors.
61
Upon payment in shares pursuant to the exercise or settlement of an award, the number of shares available for issuance under the 2025 Plan is reduced only by the number of shares actually issued in such payment. If a participant pays the exercise price (or purchase price, if applicable) of an award through the tender of shares, or if the shares are tendered or withheld to satisfy any tax withholding obligations, the number of shares so tendered or withheld again become available for issuance pursuant to future awards under the 2025 Plan, although such shares do not again become available for issuance as incentive stock options. Shares are not deemed to have been issued pursuant to the 2025 Plan with respect to any portion of an award that is settled in cash. If any outstanding award expires or is terminated or canceled without having been exercised or settled in full, or if the shares acquired pursuant to an award subject to forfeiture or repurchase are forfeited or repurchased by the Company, the shares allocable to the terminated portion of such award or such forfeited or repurchased shares again become available for grant under the 2025 Plan. No more than ten percent (10%) of the shares outstanding on December 1, 2025 (subject to adjustment pursuant to the 2025 Plan) may be issued under the 2025 Plan upon the exercise of incentive stock options.
The 2025 Plan is administered by a committee of at least one person as the Board of Directors may appoint or, if no such committee has been appointed by the Board of Directors, the 2025 Plan is administered by the Board of Directors (the “Administration Committee”). The plan administrator, which initially is the Administration Committee, has full power to select, from among the individuals eligible for awards, the individuals to whom awards will be granted, to make any combination of awards to participants, and to determine the specific terms and conditions of each award, subject to the provisions of the 2025 Plan. The plan administrator may delegate to one or more officers of the Company some or all of its authority under the 2025 Plan, including the authority to grant all types of awards to individuals who are not subject to the reporting and other provisions of Section 16 of the Exchange Act, subject to certain limitations and guidelines.
Persons eligible to participate in the 2025 Plan are officers, employees, non-employee directors, consultants, and advisors of the Company and its subsidiaries as selected from time to time by the plan administrator in its discretion. As of the date of this Annual Report, approximately sixteen individuals are eligible to participate in the 2025 Plan, which includes approximately two officers, no employees who are not officers, six non-employee directors, and one consultant/independent contractor.
Stock Options
The 2025 Plan permits the granting of stock options to officers, employees, non-employee directors, consultants, and advisors of the Company and its subsidiaries as selected from time to time by the plan administrator in its discretion. The per share exercise price for shares to be issued pursuant to exercise of a stock option is determined by the plan administrator but may be no less than 100% of the fair market value per share on the date of grant.
The exercise period of stock options is determined by the plan administrator, subject to the limitations set forth in the 2025 Plan. Upon exercise of the stock options, the exercise price must be paid by either (i) cash, (ii) check, (iii) if approved by the plan administrator, as determined in its sole discretion, surrender of other shares which meet the conditions established by the plan administrator to avoid adverse accounting consequences to the Company (as determined by the plan administrator), (iv) if approved by the plan administrator, as determined in its sole discretion, by a broker-assisted cashless exercise in accordance with procedures approved by the plan administrator, (v) if approved by the plan administrator for a nonqualified stock option, as determined in its sole discretion, by delivery of a notice of “net exercise” to the Company, pursuant to which the participant receives the number of shares underlying the stock option so exercised reduced by the number of shares equal to the aggregate exercise price of the stock option divided by the fair market value on the date of exercise, and (vi) such other method of payment permitted by applicable law.
Stock Appreciation Rights
The plan administrator may award stock appreciation rights subject to such conditions and restrictions as it may determine. Stock appreciation rights entitle the recipient to shares of common stock of the Company, or cash, equal to the value of the appreciation in the Company’s stock price over the exercise price. The exercise price generally may not be less than 100% of the fair market value of common stock of the Company on the date of grant. The plan administrator determines at what time or times each stock appreciation right may be exercised, including the ability to accelerate the vesting of such stock appreciation rights.
62
Restricted Stock and Restricted Stock Units
The plan administrator may award restricted shares of common stock of the Company and restricted stock units to participants subject to such conditions and restrictions as it may determine. These conditions and restrictions may include the achievement of certain performance goals and/or continued employment with the Company through a specified vesting period. Unless the plan administrator determines otherwise, restricted stock is held by the Company as escrow agent until the restrictions on such restricted stock have lapsed. The plan administrator, in its discretion, may accelerate the time at which any restrictions lapse or are removed. Restricted stock may be granted to participants in recognition of past services or for other valid consideration and may be issued in lieu of cash compensation due to such participant. The plan administrator may grant dividend equivalent rights to participants that entitle the recipient to receive credits for dividends that would be paid if the recipient had held a specified number of shares of common stock of the Company.
Other Stock-Based Awards
Other stock-based awards may be granted either alone, in addition to, or in tandem with, other awards granted under the 2025 Plan and/or cash awards made outside of the 2025 Plan. The plan administrator has authority to determine the participants to whom and the time or times at which other stock-based awards are made, the amount of such other stock-based awards, and all other conditions of the other stock-based awards including any dividend and/or voting rights.
Other Material Features
The 2025 Plan requires the plan administrator to make appropriate adjustments to the number of shares of common stock that are subject to the 2025 Plan, to certain limits in the 2025 Plan, and to any outstanding awards to reflect stock dividends, stock splits, extraordinary cash dividends and similar events.
In the event of a change in control, each outstanding award is assumed or an equivalent award substituted by the acquiring or successor corporation or a parent of the acquiring or successor corporation. Unless determined otherwise by the plan administrator, in the event that the successor corporation refuses to assume or substitute the award, (A) the participant fully vests in and has the right to exercise the award as to all of the shares, including those as to which it would not otherwise be vested or exercisable; (B) all applicable restrictions lapse; and (C) all performance objectives and other vesting criteria are deemed achieved at targeted levels. If a stock option or a stock appreciation right is not assumed or substituted in the event of a change in control, the plan administrator notifies the respective participant that the stock option or the stock appreciation right is exercisable, to the extent vested, for a period of up to fifteen (15) days from the date of such notice, and the stock option or stock appreciation right terminates upon the expiration of such period.
Unless determined otherwise by the plan administrator, an award may not be sold, pledged, assigned, hypothecated, transferred, or disposed of in any manner, except to the participant’s estate or legal representative, and may be exercised, during the lifetime of the participant, only by the participant, although the plan administrator, in its discretion, may permit award transfers for purposes of estate planning or charitable giving.
The Board of Directors may at any time amend, alter, suspend, or terminate the 2025 Plan. The Company may obtain stockholder approval of the 2025 Plan to the extent necessary or, as determined by the plan administrator, desirable to comply with applicable laws, including any amendment that (i) increases the number of shares available for issuance under the 2025 Plan, or (ii) changes the persons or class of persons eligible to receive awards.
All awards granted under the 2025 Plan are subject to recoupment in accordance with any clawback policy that the Company is required to adopt pursuant to the listing standards of any national securities exchange or association on which Company securities are listed or as is otherwise required by the U.S. Dodd-Frank Wall Street Reform and Consumer Protection Act or other applicable law. In addition, the Board of Directors may impose such other clawback, recovery or recoupment provisions in an award agreement as the Board of Directors determines necessary or appropriate.
No awards under the 2025 Plan have been made prior to the date of this Annual Report.
63
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth, as of May 1, 2026, certain information as to the Company’s common stock beneficially owned by persons known by the Company to own in excess of 5% of the outstanding shares of such stock. In addition, the table includes information regarding the shares of the Company’s common stock beneficially owned by (i) each named executive officer, (ii) each of the Company’s directors and (iii) the Company’s directors and executive officers as a group. Management knows of no person, except as listed below, who beneficially owned more than 5% of the outstanding shares of the Company’s common stock as of April 29. 2026. Except as otherwise indicated, the information provided in the following table was obtained from filings with the SEC and the Company pursuant to the Exchange Act. For purposes of the following table, in accordance with Rule 13d-3 under the Exchange Act, a person is deemed to be the beneficial owner of any shares of the Company’s common stock which he or she has or shares, directly or indirectly, voting or investment power, or which he or she has the right to acquire beneficial ownership of at any time within 60 days after April 29. 2026. As used herein, “voting power” is the power to vote, or direct the voting of, shares, and “investment power” includes the power to dispose of, or direct the disposition of, such shares. Unless otherwise noted, each beneficial owner has sole voting and sole investment power over the shares beneficially owned. Unless otherwise noted, the business address of each of the following entities or individuals is 100 Matawan Rd, Suite 325, Matawan, New Jersey 07747.
| Name of Beneficial Owner | Number of Shares Beneficially Owned as of Information Statement Date(8) | % of Common Stock as of Information Statement Date | Number of Shares Beneficially Owned Post Conversions and Post Split(9) | % of Common Stock Post Conversions and Post Split | ||||||||||||
| Directors and Named Executive Officers: | ||||||||||||||||
| Dr. Vin Menon(1) | — | — | 969,550 | 5.92 | % | |||||||||||
| Erick Frim | — | — | — | — | ||||||||||||
| Jakap Koo | 615,385 | 4.89 | % | 61,539 | 0.38 | % | ||||||||||
| Christopher Kim, MD. | — | — | — | — | ||||||||||||
| Bennett Weintraub, PhD | — | — | — | — | ||||||||||||
| Elona Kogan | — | — | — | — | ||||||||||||
| Carol O’Donnell | — | — | — | — | ||||||||||||
| All directors and officers as a group (7 individuals) | 615,385 | 4.89 | % | 1,031,089 | 6.36 | % | ||||||||||
| 5% or Greater Stockholders: | ||||||||||||||||
| Apimeds Inc.(2) | 4,316,618 | 34.32 | % | 431,662 | 2.64 | % | ||||||||||
| Inscobee Inc.(3) | 2,099,747 | 16.67 | % | 209,975 | 1.28 | % | ||||||||||
| Dominus IB, Inc(4) | 800,000 | 6.36 | % | 80,000 | 0.49 | % | ||||||||||
| Calfin Capital Private Limited(5) | — | — | 10,158,584 | 62.13 | % | |||||||||||
| Sea Rider Capital, LLC(6) | — | — | 969,548 | 5.93 | % | |||||||||||
| Alto Opportunity Master Fund, SPC – Segregated Master Portfolio B(7) | — | — | — | — | ||||||||||||
| (1) | Dr. Vin Menon received 484,775 shares of Preferred Stock as part of the Merger Consideration. Following the Preferred Stock Conversion, such shares of Preferred Stock will convert into 9,695,500 shares of Common Stock pursuant to the applicable conversion ratio of 20-for-1. Following the Reverse Stock Split at a ratio of 1-for-10, such stockholder will hold 969,550 shares of Common Stock. |
| (2) | Represents shares held directly by Apimeds Inc., based solely on information reported in the Schedule 13D filed with the SEC on January 26, 2026. Apimeds Inc. is a wholly owned subsidiary of Inscobee Inc. Inscobee Inc. has voting and investment control over the shares held by Apimeds Inc. Millenium Holdings is controlled by You In Soo, and as such, Mr. Yoo may be deemed to have beneficial ownership over the shares held by both Inscobee Inc. and Apimeds Inc. The business address for Apimeds Inc. is 107, Gasan Digital 2-ro, Geumcheon-gu, Seoul, Korea. The business address for Millenium Holdings is 107, Gasan Digital 2-ro, Geumcheon-gu, Seoul, Korea. Each of the parties named in this footnote disclaims any beneficial ownership of the reported shares other than to the extent of any pecuniary interest the party may have therein. |
64
| (3) | Represents shares held directly by Inscobee Inc., based solely on information reported in the Schedule 13D filed with the SEC on January 26, 2026. Millenium Holdings has voting and investment control with respect to the shares held by Inscobee Inc. Millenium Holdings is controlled by You In Soo, and as such, Mr. Yoo may be deemed to have beneficial ownership over the shares held by both Inscobee Inc. and Apimeds Inc. The business address for Inscobee Inc. is Room 613, Digital-ro 130, 6F, Geumcheon-gu, Seoul, 08580 Republic of Korea. The business address for Millenium Holdings is 107, Gasan Digital 2-ro, Geumcheon-gu, Seoul, Korea. Each of the parties named in this footnote disclaims any beneficial ownership of the reported shares other than to the extent of any pecuniary interest the party may have therein. |
| (4) | Dominus IB, Inc. is controlled by its Chief Executive Officer and largest shareholder, Park Kyoung Jin, who may be deemed to have voting and investment control with respect to the shares held by Dominus IB, Inc. The business address for Dominus IB, Inc. is 144, Dobong-ro, Gangbuk, Seoul, Republic of Korea. |
| (5) | Calfin Capital Private Limited received 5,079,292 shares of Preferred Stock as part of the Merger Consideration. Following the Preferred Stock Conversion, such shares of Preferred Stock will convert into 101,585,840 shares of Common Stock pursuant to the applicable conversion ratio of 20-for-1. Following the Reverse Stock Split at a ratio of 1-for-10, such stockholder will hold 10,158,584 shares of Common Stock. The business address for Calfin Capital Private Limited is 60 Paya Lebar Road, #04-23, Paya Lebar Square, Singapore 409051. |
| (6) | Sea Rider Capital, LLC received 484,774 shares of Preferred Stock as part of the Merger Consideration. Following the Preferred Stock Conversion, such shares of Preferred Stock will convert into 9,695,480 shares of Common Stock pursuant to the applicable conversion ratio of 20-for-1. Following the Reverse Stock Split at a ratio of 1-for-10, such stockholder will hold 969,548 shares of Common Stock. The business address for Sea Rider Capital, LLC is 850 New Burton Road, Suite 201, Dover, County of Kent, Delaware 19904, USA. |
| (7) | Based solely on information reported in the Schedule 13G filed with the SEC on February 11, 2026. Represents 1,397,021 shares of Common Stock issuable on the conversion of certain convertible notes (the “Notes”) held by the Reporting Persons. The issuable shares of Common Stock related to the conversion of the Notes are subject to a 9.99% beneficial ownership blocker. The shares reported herein represent Common Stock of the Company held by Alto Opportunity Master Fund, SPC- Segregated Master Portfolio B, a Cayman Islands exempted company (the “Fund”). The business address for the Fund is Suite #7 Grand Pavilion Commercial Centre, 802 West Bay Road, Grand Cayman, P.O. Box 10250, Cayman Islands. The Fund is a private investment vehicle for which Ayrton Capital LLC, a Delaware limited liability company (the “Investment Manager”), serves as the investment manager. The business address for the Investment Manager is 55 Post Rd West, 2nd Floor Westport, CT 06880. Waqas Khatri serves as the managing member of the Investment Manager (all of the foregoing, collectively, the “Reporting Persons”). The business address for Waqas Khatri is 55 Post Rd West, 2nd Floor Westport, CT 06880. The 1,397,021 shares of Common Stock are prior to giving effect to the Reverse Stock Split. After giving effect to the Reverse Stock Split the Fund will hold 139,703 shares of Common Stock. |
| (8) | Represents beneficial ownership of the Company’s Common Stock based on 12,575,983 shares of our Common Stock issued and outstanding as of February 26, 2026, the date of filing of this Annual Report, prior to giving effect to the Preferred Stock Conversion, the Notes Conversion, and the Reverse Stock Split. The beneficial ownership information set forth herein does not reflect any adjustments resulting from the Preferred Stock Conversion, the Notes Conversion, and the Reverse Stock Split. |
| (9) | Represents beneficial ownership of the Company’s Common Stock based on 16,351,336 shares of our Common Stock, calculated on a proforma basis, after giving effect to the Preferred Stock Conversion, the Notes Conversion, and the Reverse Stock Split. |
Changes in Control
On December 1, 2025, the Company completed the Merger with MindWave Innovations Inc. See “Item 1. Business” for a description of the Merger.
Other than the Merger, management of the Company knows of no arrangements, including any pledge by any person of securities of the Company, the operation of which may at a subsequent date result in a change in control of the registrant.
65
Item 13. Certain Relationships and Related Transactions, and Director Independence
Certain Relationships and Related Transactions
Other than as listed below, during 2025 and 2024, we were not a participant in any transaction or series of transactions in which the amount involved did exceed or may exceed the lesser of $120,000 or 1% of the average of our total assets at year-end for 2025 and 2024 in which any directors, director nominees, executive officers, greater than 5% beneficial owners and their respective immediate family members (each, a “Related Person”) had or will have a direct or indirect material interest, other than the compensation arrangements (including with respect to equity compensation) described in “Executive Compensation” beginning on page 55 and “Director Compensation” on page 56.
Business Agreement
On August 2, 2021, we entered into an agreement with Apimeds Korea, a principal stockholder of the Company (the “Business Agreement”). Pursuant to the Business Agreement, Apimeds Korea granted to the Company a sublicensable, royalty-bearing license to research, develop, manufacture and commercialize and sell Apitox in the United States. In exchange for this license, the Company will pay Apimeds Korea a perpetual royalty of 5% of the Company’s earnings before interest and taxes as determined consistent with GAAP, derived from the sale or license of Apitox, less any shipping, handling, and insurance charges, credits (arising from returns or other adjustments), discounts, rebates, or allowances of any kind (if any). The Business Agreement may be terminated by mutual written agreement by the parties and will automatically terminate upon the bankruptcy or dissolution of the Company.
Assignment Agreement
On October 12, 2021 we entered into an intellectual property assignment agreement (the “Assignment Agreement”) with Apimeds Korea and Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and founder of Apimeds Korea, effective as of May 12, 2021. Pursuant to the Assignment Agreement Dr. Kim transferred to Apimeds Korea all right, title, interest and good will in all of the intellectual property as it relates to Apitoxin, which will be marketed in the United States as Apitox (the “Assigned IP”).
Dr. Kim retained no right to use the Assigned IP. Additionally, the Assignment Agreement acknowledged that the Assigned IP was licensed to us to use via the Business Agreement, as described above.
Patent License Agreement
On October 12, 2021, we entered into a patent license agreement (the “Patent License Agreement”) Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and the founder of Apimeds Korea. During Dr. Kim’s engagement with Apimeds Korea, he contributed to the development of the intellectual property as it relates to Apitoxin. Pursuant to the Patent License Agreement, we were licensed certain patents. In consideration of its license under the Patent License Agreement, the Company paid Dr. Kim $1.00.
The patents expired in 2023 and, presently, the Company does not intend renew the expired patents or apply for any additional patents.
Business Establishment Agreement
On March 3, 2020, Apimeds Korea entered into a business establishment agreement with the Company pursuant to which Apimeds Korea agreed provide funding to us in the form of two tranches consisting of $500,000 each (for a total of $1,000,000). The first tranche was funded in March 2020 and the second tranche was funded in May 2020.
August 2021 Promissory Note
The Company issued to Apimeds Korea a convertible promissory note in the principal amount of $400,000, on August 30, 2021 (the “August 2021 Note”). The August 2021 Note is due and payable on the earlier of (i) August 30, 2026 or (ii) a sale of the Company (as defined in the August 2021 Note) (the “Maturity Date”). The August 2021 Note bears interest at an annual rate equal to the lesser of (i) 5% per annum, or (ii) the maximum rate permissible by law.
66
The Company may prepay the August 2021 Note at any time without penalty. If not previously paid by the Company, principal and accrued interest on the August 2021 Note will automatically convert into common stock (i) immediately prior to the closing of the Company’s firm commitment underwritten initial public offering resulting in at least $40,000,000 gross proceeds to the Company (a “Qualified IPO”), (ii) immediately prior to the closing of the Company’s initial listing of its common stock on an international exchange by means of an effective registration statement on Form S-1 that results in at least $40,000,000 of gross proceeds to the selling stockholders (a “Qualified Direct Listing”), or (iii) upon the consummation of the Company’s merger, consolidation, share exchange or other transaction with a publicly traded “special purpose acquisition company” resulting in a stock exchange listing (a “SPAC Transaction”). The number of shares of common stock shall be determined by dividing (x) the outstanding principal balance of the Apimeds Korea Note plus accrued but unpaid interest by (y) as applicable, (i) in case of a Qualified IPO, the per share price for which shares of common stock are initially offered in the Qualified IPO as reflected in the final prospectus, (b) in case of a Qualified Direct Listing, the fist closing price of the common stock on the first trading day, following the Qualified Direct Listing, and (c) in case of a SPAC Transaction, the price per share of the successor entity that is established in connection with such SPAC Transaction.
If there shall be any Event of Default (as defined below), the August 2021 Note shall accelerate and all principal and unpaid accrued interest shall become immediately due and payable, provided that the Company shall have 20 days from receipt of such notice to cure an Event of Default. The occurrence of any one or more of the following shall constitute an “Event of Default”: (a) the Company fails to pay timely all or any part of the principal amount or accrued interest due under the August 2021 Note, (b) the Company files any petition or action for relief under any bankruptcy, reorganization, insolvency or moratorium law or any other law for the relief of, or relating to, debtors, or makes any assignment for the benefit of creditors or takes any corporate action in furtherance of any of the foregoing, or (c) an involuntary petition is filed against the Company, or a custodian, receiver, trustee, assignee for the benefit of creditors (or other similar official) is appointed to take possession, custody or control of any property of the Company.
The terms of the August 2021 Note may only be amended with the written consent of both parties and my only be transferred upon its surrender to the Company for registration of transfer or accompanied by a duly executed written instrument of transfer in the form satisfactory to the Company.
On December 5, 2023, the Company and Apimeds Korea amended the August 2021 Note (the “August 2021 Note Amendment”) as follows: the maturity date was extended to the earlier of (i) December 31, 2026, or (ii) the consummation of an offering of our common stock (and other securities potentially) resulting in the listing for trading of our common stock on the NYSE American, the Nasdaq Capital Market, the Nasdaq Global Market, the Nasdaq Global Select Market or the New York Stock Exchange (or any successors to any of the foregoing) (“Qualified Offering”).
Additionally, the August 2021 Note Amendment provided for conversion of the note, including accrued and unpaid interest, at a conversion price of $2.60 per share as follows: (i) at the option of the holder, in its sole discretion, in whole or in part, and (ii) mandatorily simultaneous with the consummation of a Qualified Offering, in each case, into fully paid and nonassessable shares of common stock at the conversion price.
On June 12, 2024, Apimeds Korea assigned the August 2021 Note to Inscobee, Inc. a South Korean company, and the parent company of Apimeds Korea, (“Inscobee”).
If not converted earlier, upon the closing of a Qualified Offering, the August 2021 Note will automatically convert into approximately 179,283 shares of common stock.
March 2022 Promissory Note
The Company issued to Apimeds Korea, a promissory note in the principal amount of $160,000 on March 21, 2022 (the “March 2022 Note”). The March 2022 Note bears interest at a rate equal to 5% per annum (the “Interest Rate”). The March 2022 Note is due and payable on the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000, or (ii) July 15, 2022.
The Company may prepay the March 2022 Note at any time without penalty. If any payment due on the March 2022 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and Inscobee may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
67
On December 5, 2023, the Company and Apimeds Korea amended the March 2022 Note (the “March 2022 Note Amendment”) as follows: the maturity date was extended to the earlier of (i) December 31, 2026, or (ii) the consummation of a Qualified Offering.
Additionally, the March 2022 Note Amendment provided for conversion of the note, including accrued and unpaid interest, at a conversion price of $2.60 per share as follows: (i) at the option of the holder, in its sole discretion, in whole or in part, and (ii) mandatorily simultaneous with the consummation of a Qualified Offering, in each case, into fully paid and nonassessable shares of common stock at the conversion price.
On June 12, 2024, Apimeds Korea assigned the March 2022 Note to Inscobee.
If not converted earlier, upon the closing of a Qualified Offering, the March 2022 Note will automatically convert into approximately 70,002 shares of common stock.
June 2022 Promissory Note
On June 3, 2022, the Company issued to Inscobee, Inc. a South Korean company, and the parent company of Apimeds Korea, (“Inscobee”) a $100,000 promissory note (the “June 2022 Note”). Interest on the outstanding principal balance of the Second Loan accrues at a rate equal to 5% per annum, and interest on the outstanding principal balance of the First Loan shall accrue and be payable on the maturity date. The maturity date was the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000), and (ii) July 15, 2022.
On December 5, 2023, the Company and Inscobee amended the June 2022 Note (the “June 2022 Note Amendment”) as follows: the maturity date was extended to (i) December 31, 2026, or (ii) consummation of a Qualified Offering.
Additionally, the June 2022 Note Amendment provided for conversion of the note, including accrued and unpaid interest, at a conversion price of $2.60 per share as follows: (i) at the option of the holder, in its sole discretion, in whole or in part, and (ii) mandatorily simultaneous with the consummation of a Qualified Offering, in each case, into fully paid and nonassessable shares of common stock at the conversion price.
Upon the closing of a Qualified Offering, the June 2022 Note will automatically convert into approximately 43,361 shares of common stock.
On June 12, 2024, the Company and Inscobee amended the June 2022 Note to correct a scrivener’s error.
May 2024 Promissory Note
On May 20, 2024, the Company issued to Inscobee a $100,000 promissory note (the “May 2024 Note”). The May 2024 Note bears interest at a rate equal to 5% per annum (the “Interest Rate”). The May 2024 Note is due and payable on the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000, or (ii) May 19, 2025.
The Company may prepay the May 2024 Note at any time without penalty. If any payment due on the May 2024 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and Inscobee may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
August 2024 Note
On August 19, 2024, the Company issued to Inscobee a $150,000 principal amount promissory note (the “August 2024 Note”). The August 2024 Note bears interest at a rate equal to 5% per annum (the “Interest Rate”). The August 2024 Note is due and payable on the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000, or (ii) May 19, 2025. The outstanding principal and interest on the August 2024 Note will be repaid upon the closing of a Qualified Offering.
68
The Company may prepay the August 2024 Note at any time without penalty. If any payment due on the August 2024 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and Inscobee may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
March 2025 Promissory Note
On March 31, 2025, the Company received $250,000 in a promissory note (the “March 2025 Note”) agreement with Apimeds, Inc., one of its shareholders. The Promissory Notes bear interest at 5% per annum and mature on the earlier of (a) December 31, 2026 or (b) consummation of a Qualified Offering (the “Maturity Date”). “Qualified Offering” shall mean an offering of Common Stock (and other securities potentially) resulting in the listing for trading of the Common Stock on the NYSE American, the Nasdaq Capital Market, the Nasdaq Global Market, the Nasdaq Global Select Market or the New York Stock Exchange (or any successors to any of the foregoing).
The Company may prepay the March 2025 Note at any time without penalty. If any payment due on the March 2025 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
Cash Advance Loans
On October 5, 2022, November 10, 2022 and March 16, 2023, Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and founder of Apimeds Korea, loaned the Company $9,900, $13,000 and $9,000 respectively. These loans carried no interest and did not have a maturity date. The loans were used for operating purposes. As of September 2023, all the loan amounts were repaid.
Policies and Procedures for Transaction with Related Persons
It is the responsibility of our audit committee to review and approval all related party transactions that would need to be disclosed pursuant to Item 404(a) of Regulation S-K (each a “Related Party Transaction”). The Board has adopted a related party transaction policy that makes up a part of the audit committee’s charter (the “Related Party Transactions Policy”). Pursuant to the Related Party Transactions Policy, each of the Company’s directors and executive officers shall promptly inform the chairperson of the audit committee of any potential Related Party Transactions. In addition, each such director and executive officer shall complete a questionnaire on an annual basis designed to elicit information about any potential Related Party Transactions. Any potential Related Party Transactions that are brought to the audit committee’s attention shall be analyzed by the audit committee, in consultation with outside counsel or members of management, as appropriate, to determine whether the transaction or relationship does, in fact, constitute a Related Party Transaction requiring compliance with the Related Party Transactions Policy. In determining whether to approve a Related Party Transaction, the audit committee shall consider, among other factors, the following factors to the extent relevant to the Related Party Transaction: (i) whether the terms of the Related Party Transaction are fair to the Company and on the same basis as would apply if the transaction did not involve a Related Party (as defined in the Related Party Transactions Policy); (ii) whether there are business reasons for the Company to enter into the Related Party Transaction; (iii) whether the Related Party Transaction would impair the independence of an outside director; (iv) whether the Related Party Transaction would present an improper conflict of interest for any director or executive officer of the Company, taking into account the size of the transaction, the overall financial position of the director, executive officer or Related Party, the direct or indirect nature of the director’s, executive officer’s or Related Party’s interest in the transaction and the ongoing nature of any proposed relationship, and any other factors the Committee deems relevant; and (v) any pre-existing contractual obligations. All of the transactions described in this section occurred prior to the adoption of this policy.
69
Director Independence
The Company’s Board has determined that Dr. Bennett Weintraub, PhD, Carol O’Donnell, and Elona Kogan, who together comprise a majority of the Board, are independent under applicable rules and regulations of the SEC. The Board made such independence determinations using the definition of independence set forth in the rules of the NYSE American based on a review of transactions and relationships between each director or any member of his or her immediate family, on the one hand, and the Company and its subsidiaries and affiliates, on the other hand, as well as transactions and relationships between each director or his affiliates, on the one hand, and members of the Company’s management or their affiliates, on the other hand.
Item 14. Principal Accountant Fees and Services
Kreit & Chiu CPA LLP served as the independent registered public accounting firm for the Company for 2025 and 2024. The following table sets forth the fees billed to the Company by Kreit & Chiu CPA LLP for 2025 and 2024.
| 2025 | 2024 | |||||||
| (in thousands) | ||||||||
| Audit Fees (1) | $ | 206,933 | $ | 142,918 | ||||
| Audit-Related Fees | - | |||||||
| All Other Fees | - | - | ||||||
| Total Fees | $ | 206,933 | $ | 142,918 | ||||
Under its charter, the Company’s audit committee must review and pre-approve both audit and permitted non-audit services provided by the Company’s independent registered public accounting firm and shall not engage the independent registered public accounting firm to perform any non-audit services prohibited by law or regulation. The independent registered public accounting firm’s retention to audit the Company’s financial statements, including the associated fee, is subject to approval each year by the audit committee. The audit committee does not regularly evaluate potential engagements of the independent registered public accounting firm and approve or reject such potential engagements. At each audit committee meeting, the audit committee receives updates on the services actually provided by the independent registered public accounting firm, and management may present additional services for pre-approval. The audit committee may delegate to the chairman of the audit committee the authority to evaluate and approve engagements on behalf of the audit committee in the event that a need arises for pre-approval between regular audit committee meetings. If the chairman so approves any such engagements, he will report that approval to the full audit committee at the next audit committee meeting.
The audit committee was established on February 7, 2025. Accordingly, audit and audit-related services rendered prior to February 7, 2025 were not pre-approved by the audit committee. All audit and audit-related services rendered after February 7, 2025, including services related to the audit of the Company’s financial statements for the fiscal year ended December 31, 2025, were pre-approved by the audit committee in accordance with the foregoing procedures.
70
PART IV
Item 15. Exhibit and Financial Statement Schedules
(a) Documents filed as part of this report
(1) All financial statements
(2) Financial Statement Schedules
All financial statement schedules are omitted because they are either inapplicable or not required, or because the required information is included in the Financial Statements or notes thereto contained in this Annual Report on Form 10-K.
(3) Exhibits required by Item 601 of Regulation S-K
71
| 21.1* | Subsidiaries of the Registrant. | |
| 31.1* | Certification of Chief Executive Officer Pursuant to Securities Exchange Act Rules 13a-14(a), as adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2* | Certification of Chief Financial Officer Pursuant to Securities Exchange Act Rules 13a-14(a), as adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1* | Certification of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2* | Certification of Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 97.1* | Executive Compensation Recovery Policy. | |
| 101.INS | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |
| 101.LAB | Inline XBRL Taxonomy Extension Labels Linkbase Document. | |
| 101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |
| 104 | Cover Page Interactive Data File (Embedded within the Inline XBRL document and included in Exhibit). | |
| 101.INS | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document. |
| * | Filed or furnished herewith. |
Item 16. Form 10-K Summary
None.
72
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| APIMEDS PharmaCEUTICALS US, Inc. | ||
| Date: May 3, 2026 | /s/ Dr. Vin Menon | |
| Name: | Dr. Vin Menon | |
| Title: | Chief Executive Officer | |
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
| SIGNATURE | TITLE | DATE | ||
| /s/ Dr. Vin Menon | Chief Executive Officer | May 3, 2026 | ||
| Dr. Vin Menon | (Principal Executive Officer) | |||
| /s/ Erick Frim | Chief Financial Officer | May 3, 2026 | ||
| Erick Frim | (Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Dr. Bennett Weintraub, PhD | Director | May 3, 2026 | ||
| Dr. Bennett Weintraub, PhD | ||||
| /s/ Carol O’Donnell | Director | May 3, 2026 | ||
| Carol O’Donnell | ||||
| /s/ Elona Kogan | Director | May 3, 2026 | ||
| Elona Kogan |
73
INDEX TO FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
Board of Directors and Shareholders
Apimeds Pharmaceuticals US, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Apimeds Pharmaceuticals US, Inc. (the “Company”) as of December 31, 2025, and 2024, and the related consolidated statements of operations, statements of changes in shareholders’ equity (deficit), and cash flows for each of the two years in the period ended December 31, 2025, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2025 and 2024, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2025, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 3 to the consolidated financial statements, the Company has suffered recurring losses from operations and negative cash flows from operations which raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 3. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the entity’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the entity’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
We have served as the Company’s auditor since 2023.
May 3, 2026
F-2
| Apimeds Pharmaceuticals US, Inc. |
| Consolidated Balance Sheets |
| December 31, | December 31, | |||||||
| 2025 | 2024 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash & Cash Equivalents | $ | $ | ||||||
| Restricted Cash | ||||||||
| Short Term Investments | ||||||||
| Prepaid Expenses | ||||||||
| Other Current Assets | ||||||||
| Total current assets | ||||||||
| Digital assets, at fair value | ||||||||
| Long-term portion of prepaid expenses | ||||||||
| Operating Lease ROU Asset | ||||||||
| Property and Equipment, net | ||||||||
| Total assets | $ | $ | ||||||
| Liabilities and shareholders’ equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Accrued offering costs | ||||||||
| Accrued interest - related party | ||||||||
| Advance payable to related party | ||||||||
| Notes payable - related party | ||||||||
| Derivative Liability | ||||||||
| Convertible Notes, net | ||||||||
| Operating Lease Liability | ||||||||
| Total current liabilities | ||||||||
| Long-term liabilities | ||||||||
| Long-Term Portion of Operating Lease Liability | ||||||||
| Long-term convertible notes payable – related party | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies | ||||||||
| Shareholders’ equity: | ||||||||
| Common stock, par value $ | ||||||||
| Additional paid-in capital | ||||||||
| Retained Earnings (Deficit) | ( | ) | ( | ) | ||||
| Total shareholders’ equity (deficit) | ( | ) | ||||||
| Total liabilities and shareholders’ equity | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Apimeds Pharmaceuticals US, Inc.
Consolidated Statements of Operations
| For the year ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Operating expenses: | ||||||||
| Research and development expenses | $ | $ | ||||||
| General and administrative expenses | ||||||||
| Total operating expenses | ||||||||
| Loss from operations | ( | ) | ( | ) | ||||
| Other income (expense) | ||||||||
| Unrealized gain from changes in fair value of digital assets | ||||||||
| Realized gain on sale of digital assets | ||||||||
| Trading gains, net | ||||||||
| Foreign currency gains/(losses), net | ( | ) | ||||||
| Change in FV of warrant liability | ||||||||
| Change in FV of derivative | ||||||||
| Interest income | ||||||||
| Interest expense | ( | ) | ( | ) | ||||
| Total other income (expense) | ( | ) | ||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Net loss per common share - basic and diluted | $ | ( | ) | $ | ( | ) | ||
| Weighted average common shares outstanding | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
Apimeds Pharmaceuticals US, Inc.
Consolidated Statement of Changes in Shareholders Equity (Deficit)
| Preferred Stock | Common Stock | Additional | ||||||||||||||||||||||||||
| Number of Shares | Amount | Number of Shares | Amount | Paid-in capital | Accumulated Deficit | Total | ||||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||||||
| Net loss for the period ended December 31, 2024 | - | - | ( | ) | ( | ) | ||||||||||||||||||||||
| Balance at December 31, 2024 | - | ( | ) | ( | ) | |||||||||||||||||||||||
| Stock-based compensation - stock options | - | - | ||||||||||||||||||||||||||
| Stock-based compensation – common stock grants | - | |||||||||||||||||||||||||||
| Conversion of convertible debt - related party | - | |||||||||||||||||||||||||||
| Issuance of Representative Warrants in connection with IPO | - | - | ||||||||||||||||||||||||||
| Issuance of common stock in IPO (net of $ | - | |||||||||||||||||||||||||||
| Issuance of Representative Warrants in connection with IPO | - | - | ||||||||||||||||||||||||||
| Issuance of Advisory Warrants in connection with Merger | - | - | ||||||||||||||||||||||||||
| Issuance of preferred stock in Merger | ||||||||||||||||||||||||||||
| Net loss for the period ended December 31, 2025 | - | - | ( | ) | ( | ) | ||||||||||||||||||||||
| Balance at December 31, 2025 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Apimeds Pharmaceuticals US, Inc.
Consolidated Statements of Cash Flows
| For the years ended December 31 | ||||||||
| 2025 | 2024 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock based compensation - Common Stock Grants | ||||||||
| Stock based compensation - stock options and warrants | ||||||||
| Change in fair value of warrant liability | ( | ) | ||||||
| Change in fair value of derivative liability | ( | ) | ||||||
| Depreciation expense | ||||||||
| Interest expense | ||||||||
| Unrealized gain from changes in fair value of digital assets | ( | ) | ||||||
| Non-cash digital asset operating expenses | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | ( | ) | ||||||
| Accounts payable and accrued expenses | ( | ) | ||||||
| Net cash used in operating activities | ( | ) | ( | ) | ||||
| Cash flows from investing activities: | ||||||||
| Cash from short term investments | ( | ) | ||||||
| Restricted cash | ( | ) | ||||||
| Purchases of PP&E | ( | ) | ||||||
| Cash paid to acquire operating lease | ( | ) | ||||||
| Cash acquired in accordance with merger | ||||||||
| Net cash used in investing activities | ( | ) | ||||||
F-6
Apimeds Pharmaceuticals US, Inc.
Consolidated Statements of Cash Flows
(continued)
| For the years ended December 31 | ||||||||
| 2025 | 2024 | |||||||
| Cash flows from financing activities: | ||||||||
| Cash proceeds from issuance of common stock in IPO | ||||||||
| Proceeds from notes payable - related parties | ||||||||
| Proceeds from PIPE convertible note | ||||||||
| Payment of debt issuance costs | ( |
) | ||||||
| Cash advances from related parties | ||||||||
| Cash advances paid to related parties | ( |
) | ||||||
| Net cash provided by financing activities | ||||||||
| Net increase (decrease) in cash, cash equivalents | ( |
) | ||||||
| Cash, cash equivalents, beginning of period | ||||||||
| Restricted cash | ||||||||
| Cash, cash equivalents, and restricted cash, end of period | $ | $ | ||||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for interest | $ | $ | ||||||
| Cash paid for taxes | $ | |||||||
| Non-cash investing and financing activities: | ||||||||
| Net Assets Acquired in the Merger | ||||||||
| Operating Lease | ( |
) | ||||||
| Reconciliation of cash, cash equivalents, and restricted cash: | ||||||||
| Cash and cash equivalents | ||||||||
| Restricted cash | ||||||||
| Total cash, cash equivalents, and restricted cash | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements
F-7
Note 1 ORGANIZATION AND DESCRIPTION OF BUSINESS
Apimeds Pharmaceuticals US, Inc. (“APUS” or the “Company”) is a development-stage biopharmaceutical company incorporated in the State of Delaware as a C-Corporation. The Company is focused on the development of Apitox, a purified honeybee venom-based drug for the treatment of acute pain and inflammation associated with knee osteoarthritis. On December 1, 2025, the Company completed a merger (the “Merger”) with MindWave Innovations Inc. (“MindWave”), whereby MindWave became a wholly owned subsidiary of the Company (see Note 4). In connection with the Merger, the Company acquired digital assets, including Bitcoin (“BTC”), Tether (“USDT”), and MindWaveDAO NILA tokens (“NILA Tokens”), and assumed certain operations related to digital asset activities.
The Company operates its biopharmaceutical business through Lokahi Therapeutics Inc. (“Lokahi”), a wholly owned subsidiary. As of December 31, 2025, the Company’s corporate structure is as follows:
| ● | APUS — Public parent and SEC registrant (Delaware C-Corporation) |
| o | Lokahi Therapeutics Inc. (“The BioBusiness”) : Wholly owned subsidiary; operates the BioBusiness segment |
| o | MindWave Innovations Inc.: (acquired December 1, 2025); operates the digital asset segment |
The Company has not yet generated revenue from its biopharmaceutical operations and is subject to the risks and uncertainties common to development-stage companies in the biotechnology industry.
Note 2 BASIS OF PRESENTATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
(a) Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and pursuant to the rules and regulations of the U.S. Securities and Exchange Commission (“SEC”). The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries, Lokahi Therapeutics Inc. and effective December 1, 2025, MindWave Innovations Inc. All intercompany balances and transactions have been eliminated in consolidation. The results of operations MindWave are included in the consolidated financial statements from the date of acquisition, December 1, 2025, through December 31, 2025. Prior-period amounts reflect the operations of the BioBusiness only.
(b) Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Significant estimates include, but are not limited to, the fair value of consideration transferred and net assets acquired in the Merger, the fair value of digital assets, the fair value of stock-based compensation awards, the fair value of warrants, the valuation allowance on deferred tax assets, and the assessment of the Company’s ability to continue as a going concern. Actual results could differ materially from those estimates.
(c) Segment Reporting
In accordance with ASC 280, Segment Reporting, the Company has determined
that it operates as two segments: (i) the BioBusiness segment, which advances the Company’s lead product candidate, Apitox, and related
preclinical and translational research activities; and (ii) the Digital Assets segment, which encompasses the Company’s digital asset
holdings (Bitcoin, Tether, and NILA Tokens) acquired in connection with the MindWave acquisition and the activities associated with the
MindWaveDAO ecosystem.
F-8
The following tables present the Company’s segmented results for the years
ended December 31, 2025. The Company has
For the Year ended December 31, 2025
| BioBusiness Segment | Digital Asset Segment | Corporate | Consolidated | |||||||||||||
| Revenue | $ | $ | $ | $ | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | ||||||||||||||||
| General and administrative | ||||||||||||||||
| Total operating expenses | $ | $ | $ | $ | ||||||||||||
| Loss from operations | ( | ) | ( | ) | ( | ) | ||||||||||
| Other income (expense), net: | ||||||||||||||||
| Realized gain on sale of digital assets | ||||||||||||||||
| Trading gains, net | ||||||||||||||||
| Unrealized gain on digital assets | ||||||||||||||||
| Change in FV of derivative | ||||||||||||||||
| Change in FV of warrant liability | ||||||||||||||||
| Interest income | ||||||||||||||||
| Interest expense | ( | ) | ( | ) | ||||||||||||
| Foreign currency transaction loss | ( | ) | ( | ) | ||||||||||||
| Total other income (expense), net | $ | $ | $ | ( | ) | $ | ||||||||||
| Net loss | $ | ( | ) | $ | $ | ( | ) | $ | ( | ) | ||||||
(d) Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of three months or less at the date of acquisition to be cash equivalents. Cash and cash equivalents consist primarily of amounts held in demand deposit accounts.
(e) Digital Assets
Digital assets consist of Bitcoin (“BTC”), Tether (“USDT”), and MindWaveDAO NILA tokens (“NILA Tokens”). Effective upon the adoption of ASU 2023-08, Accounting for and Disclosure of Crypto Assets, the Company accounts for in-scope crypto assets that meet the definition of an intangible asset and are fungible as follows:
| ● | BTC — Measured at fair value with changes in fair value recognized in the consolidated statement of operations within “Unrealized gain (loss) on digital assets.” BTC meets the criteria of ASU 2023-08 and is classified within Level 1 of the fair value hierarchy based on quoted prices in active markets. | |
| ● | USDT — Tether is a stablecoin pegged to the U.S. dollar. The Company measures USDT at fair value and classifies USDT within Level 1 of the fair value hierarchy based on quoted prices on active cryptocurrency exchanges. Because USDT is designed to maintain a stable value relative to the U.S. dollar, changes in fair value are generally not material. | |
| ● | NILA Tokens — The NILA Tokens are utility tokens issued within the MindWaveDAO ecosystem. NILA Tokens trade on a limited number of centralized cryptocurrency exchanges, primarily the NILA/USDT trading pair. The Company measures NILA Tokens at fair value and classifies them within Level 2 of the fair value hierarchy based on quoted prices for identical or similar assets in markets that are not considered active due to the limited number of trading venues and relatively low trading volume. Gains and losses realized upon the sale of NILA Tokens are recognized within “Realized gain (loss) on sale of digital assets” in the consolidated statement of operations. Unsold NILA Tokens are remeasured at fair value at each reporting date, with unrealized changes recognized within “Unrealized gain (loss) on digital assets” in the consolidated statement of operations. |
F-9
The Company is not a broker-dealer, exchange, or investment company. Digital asset activities are limited to the holding, sale, and conversion of tokens acquired in the Merger accordingly, the digital assets are classified as noncurrent in the accompanying consolidated balance sheet.
(f) Fair Value Measurements
The Company follows a three-level hierarchy for fair value measurements as follows:
Level 1 Quoted prices in active markets for identical assets or liabilities that the Company can access at the measurement date
Level 2 Observable inputs other than quoted prices in Level 1
Level 3 Unobservable inputs requiring management estimates
(g) Mergers and Acquisitions
The Company accounts for mergers and acquisitions in accordance with ASC 805, Business Combinations. Under this standard, the Company determines whether the acquiree meets the requirements to constitute a business in accordance with ASC 805-10-55-5A. If the acquiree meets the requirements to constitute a business, the acquisition method of accounting is applied, whereby the results of operations of acquired businesses are included in the consolidated financial statements from the date of acquisition. The identifiable assets acquired and liabilities assumed are recognized at their estimated fair value at the acquisition date. Regarding the merger transaction dated December 1, 2025, the acquiree, MindWave Innovations Inc, maintains substantially all fair value of gross assets in a singular identifiable asset category. Therefore, the transaction is accounted for as an asset acquisition wherein the fair value of the net assets assumed by the company was determined to be equivalent to the Series A Convertible Preferred Stock issued as consideration. In accordance with an asset acquisition, no goodwill was recorded (See note 4).
(h) Stock-Based Compensation
The Company accounts for stock-based compensation using the fair value of equity awards measured at the grant date and recognized as expense over the requisite service period. Stock-based compensation issued to the employees of subsidiaries is recognized as a capital contribution from the Company to the respective subsidiaries (See note 8).
(i) Warrants
The Company classifies warrants as either equity or liabilities. Warrants that are indexed to the Company’s own stock and meet the criteria for equity classification are recorded in stockholders’ equity. Warrants previously classified as liabilities are measured at fair value each reporting period, with changes recognized within “Change in fair value of warrant liabilities” in the consolidated statement of operations.
(j) Revenue Recognition
The Company has not generated revenue from its biopharmaceutical operations. Proceeds from the sale of cryptocurrencies maintained by the Company, inclusive of USDT; BTC; and NILA Tokens, are recognized as realized gains or losses on sale of digital assets and are not considered revenue.
F-10
(k) Leases
The Company classifies its leases as either operating or financing. For operating leases with terms greater than 12 months, at the commencement date, the Company recognizes a right-of-use (“ROU”) asset and a corresponding lease liability. The lease liability is measured at the present value of future lease payments, discounted using the Company’s incremental borrowing rate when the rate implicit in the lease is not readily determinable. For finance leases, the Company will recognize an asset as property and equipment and a corresponding liability.
(l) Income Taxes
The Company accounts for income taxes using the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax effects of temporary differences between the financial statement carrying amounts and the tax bases of assets and liabilities. A valuation allowance is established when it is more likely than not that some or all of the deferred tax assets will not be realized.
(m) Loss Per Share
Basic loss per share is computed by dividing net loss attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period. Diluted loss per share is computed similarly, except that the denominator includes potentially dilutive securities when their effect is dilutive. For all periods presented, diluted loss per share is the same as basic loss per share because the Company was in a net loss position, and the inclusion of potentially dilutive securities would be anti-dilutive.
(n) Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist primarily of cash deposits and digital assets. Cash is maintained at financial institutions in amounts that may exceed federally insured limits. The Company has not experienced any losses on such accounts.
(o) Deferred Offering Costs
Costs directly attributable to a proposed offering of equity securities are deferred and recorded as an asset and charged against the proceeds of the offering. In the event debt offering costs are deferred, the unamortized balance is reflected as a debt discount to the principal value of the debt issued. If the offering is abandoned, deferred offering costs are charged to expense (See note 7).
Note 3 GOING CONCERN
The accompanying consolidated financial statements have been prepared
assuming the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities
in the normal course of business. Since inception, the Company has incurred recurring operating losses and negative cash flows from operations.
For the year ended December 31, 2025, the Company reported a net loss of $
F-11
Note 4 ACQUISITION OF MINDWAVE INNOVATIONS
Description of the Transaction
On December 1, 2025, Apimeds Pharmaceuticals US, Inc, a Delaware corporation (the Company or “Apimeds”), entered into an Agreement and Plan of Merger (the “Merger Agreement”) with (i) Apimeds Merger Sub, Inc., a Delaware corporation, (ii) MindWave Innovations Inc, a Delaware corporation (“MindWave”), (iii) Lokahi Therapeutics, Inc., a Nevada corporation, and (iv) Erik Emerson, solely in his capacity as representative for the BioBusiness. Unless otherwise defined herein, the capitalized terms used below are defined in the Merger Agreement. On December 10, 2025, the Merger Agreement was amended to correct scrivener’s errors in the original document.
Pursuant to the terms and conditions of the Merger Agreement, a certificate of merger (the “Certificate of Merger”) was filed with the Secretary of State of the State of Delaware (the “DE SOS”) (such time of the filing of the Certificate of Merger, the “Effective Time”), in accordance with the General Corporation Law of the State of Delaware (the “DGCL”). Pursuant to the Certificate of Merger, Merger Sub was merged with and into the MindWave (the “Merger”), with the MindWave surviving the Merger as the Surviving Corporation. As a result of the Merger, the Company MindWave became a direct wholly owned subsidiary of the Company. At the Effective Time, all the property, rights, privileges, powers and franchises of MindWave and Merger Sub were vested in Apimeds Pharmaceuticals US, the Surviving Corporation, and all the debts, liabilities and duties of the MindWave and Merger Sub became the debts, liabilities and duties of the Surviving Corporation. The Closing occurred simultaneously with the execution and delivery of the Merger Agreement on the Closing Date.
The Company determined that it is the accounting acquirer in the transaction as the consideration issued does not provide MindWave with voting control of the combined entity, MindWave does not have the ability to appoint members to the Company’s board of directors, and the senior management of the Company remains largely intact following the transaction.
In applying the concentration test practical expedient under ASC 805-10-55-5A,
the Company determined that substantially all the fair value of MindWave’s gross assets, approximately
The assets and liabilities of MindWave at the acquisition date were determined to approximate fair value, as the digital assets held by MindWave are measured at fair value and remeasured each reporting period through earnings, and the remaining assets and liabilities are short-term in nature such that their carrying values approximate fair value.
Consideration Transferred
As the transaction was consummated through the issuance of non-cash consideration in the form of Series A Convertible Preferred Stock, which is not publicly traded and does not have a readily determinable market value, the Company measured the transaction using the more readily determinable fair value of the net assets acquired in accordance with ASC 805-50-30-2.
F-12
Assets Acquired and Liabilities Assumed
The following table summarizes the fair values of the identifiable assets acquired and liabilities assumed as of the Closing Date. Because the transaction was deemed an asset acquisition, as seen in “Description of the Transaction” no goodwill was recorded.
| Fair Value at December 1, 2025 | ||||
| Assets acquired: | ||||
| Cash and cash equivalents | $ | |||
| Digital assets: BTC | ||||
| Digital assets: USDT | ||||
| Digital assets: NILA Tokens | ||||
| Other assets | ||||
| Total identifiable assets acquired | ||||
| Liabilities assumed: | ||||
| Accounts payable and accrued liabilities | $ | |||
| Other liabilities | ||||
| Total liabilities assumed | ||||
| Net identifiable assets acquired | ||||
| Goodwill | ||||
| Total consideration transferred | $ | |||
The allocation of the purchase price is based on management’s assessment of the fair values of the identifiable assets acquired and liabilities assumed as of the Closing Date.
Non-Cash Consideration Disclosure
Pursuant to the terms and conditions of the Merger Agreement, Series A Convertible Preferred Stock was issued to the respective owners of MindWave Innovations. Being a non-cash transaction, the company used the fair value of the net assets acquired by the company at the closing date of the merger, December 1, 2025, to measure the consideration as the fair value of the assets was readily determinable.
The issuance of
The digital assets acquired in the Merger with an aggregate fair value
of $
Note 5 DIGITAL ASSETS AND TOKEN ACTIVITIES
As a result of the Merger, the Company acquired digital assets consisting of BTC, USDT, and NILA Tokens on December 1, 2025. The Company holds digital assets primarily for liquidity and volatility mitigation purposes and is not a broker-dealer, exchange, or investment company.
F-13
Digital Asset Holdings
Activity During the Period (December 1, 2025, through December 31, 2025)
| BTC | USDT | NILA Tokens | Total | |||||||||||||
| Balance acquired in Merger (12/1/2025) | $ | |||||||||||||||
| Tokens sold (NILA for USDT) | $ | ( | ) | ( | ) | |||||||||||
| Realized gain (loss) on sale of NILA Tokens | $ | |||||||||||||||
| Unrealized gain (loss) recognized | $ | ( | ) | |||||||||||||
| Balance at December 31, 2025 | $ | |||||||||||||||
Realized Gains and Losses
During the period from December 1, 2025, through December 31, 2025, the
Company sold
Unrealized Gains and Losses
For the period from December 1, 2025, through December 31, 2025, the Company
recognized unrealized gains (losses) on digital assets of $
Custody and Risks
The Company is exposed to risks inherent in digital asset holdings, including price volatility, cybersecurity risks, regulatory uncertainty, and concentration risk. The Company does not insure its digital asset holdings against loss or theft. USDT is a stablecoin that seeks to maintain a 1:1 peg with the U.S. dollar. The Company is exposed to the risk that USDT could trade materially below its pegged value, that the USDT issuer (Tether Limited) may not maintain adequate reserves, or that regulatory actions could affect the redeemability or value of USDT.
NILA Tokens trade on a limited number of centralized cryptocurrency exchanges. The Company is exposed to liquidity risk, as there can be no assurance that the Company will be able to sell NILA Tokens at prices equal to or exceeding their carrying value. Daily trading volumes for NILA Tokens have historically been modest, and significant sales by the Company could adversely affect the trading price.
Cash Flow Statement Presentation
Proceeds from sales of digital assets are presented within investing activities in the consolidated statement of cash flows. Non-cash changes in the fair value of digital assets are reconciling items in the operating activities section.
FAIR VALUE MEASUREMENTS
Assets and Liabilities Measured at Fair Value on a Recurring Basis
As of December 31, 2025:
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Assets: | ||||||||||||||||
| Digital assets — BTC | $ | |||||||||||||||
| Digital assets — USDT | $ | |||||||||||||||
| Digital assets — NILA Tokens | $ | |||||||||||||||
| Total assets at fair value | $ | |||||||||||||||
| Liabilities: | ||||||||||||||||
| Warrant liabilities | ||||||||||||||||
| Derivative liability | $ | |||||||||||||||
| Total liabilities at fair value | $ | |||||||||||||||
F-14
The warrant liability as of May 12, 2025 (IPO date),
was valued utilizing the Black-Scholes options pricing model with the following inputs: $
Level 2 Valuation — NILA Tokens
NILA Tokens are classified within Level 2 of the fair value hierarchy.
The fair value of NILA Tokens is determined using quoted prices for the NILA/USDT trading pair on centralized cryptocurrency exchanges.
Although NILA Tokens have observable pricing on exchanges including LBank, the limited number of trading venues (two exchanges as of December
31, 2025) and relatively modest daily trading volume (ranging from approximately $
Level 3 Valuation — Derivative Liability
The derivative liability associated with the variable conversion feature
of the PIPE convertible note is classified within Level 3 of the fair value hierarchy. The fair value is determined using a Monte Carlo
simulation wherein a probability-weighted scenario analysis incorporating the Company’s stock price, expected volatility, remaining term
of the note, and the contractual
Level 3 Roll forward — Derivative Liability
| Beginning Balance - December 31, 2024 | $ | |||
| Initial Recognition upon Issuance | ||||
| Re-measurement adjustments: | ||||
| Change in fair value of derivative liability | ( | ) | ||
| Ending balance – December 31, 2025 | $ |
The change in fair value of derivative liability recognized in earnings
during the year ended December 31, 2025, was $
Level 3 Roll forward —
| Beginning Balance - December 31, 2024 | $ | |||
| Advisor warrant liability incurred in connection with the IPO | ||||
| Re-measurement adjustments: | ||||
| Change in fair value of warrant liability | ( | ) | ||
| Re-classification to equity upon issuance of warrants | ( | ) | ||
| Ending balance – December 31, 2025 | $ |
The change in fair value of warrant liabilities recognized in earnings
during the year ended December 31, 2025 (prior to reclassification) was $
F-15
Note 6 DEBT AND FINANCING ARRANGEMENTS
Convertible Notes
In connection with the merger, the company executed a securities purchase
agreement on December 1, 2025, of an aggregate maximum principal amount equal to $
On the date December 8, 2025, the company executed the first, and only,
senior secured convertible note (“the note”) in connection with the securities purchase agreement disclosed herein
for a principal amount of $
Related Party Notes
On the date March 21, 2025, the Company was issued
an unsecured promissory note in the principal amount of $
At December 31, 2025, the company had an advance payable to its CEO of $
Note 7 STOCKHOLDERS’ EQUITY
Authorized Capital
As of December 31, 2025, the Company’s authorized capital stock consisted of:
| Class | Shares Authorized | Par Value | ||||||
| Common Stock | ||||||||
| Preferred Stock | ||||||||
Of the authorized preferred stock,
Common Stock
As of December 31, 2025, and the subsequent period thereafter, there were
F-16
Series A Convertible Preferred Stock
In connection with the Merger (see Note 4), on December 1, 2025, the Company
issued
| ● | Conversion: Each share of Series A Preferred Stock is convertible into |
| ● | Voting rights: The Series A Preferred Stock does not maintain any voting rights. |
| ● | Redemption: The Preferred Stock issued is not redeemable |
The Company evaluated the Series A Preferred Stock under ASC 480 and determined
that the instrument is classified in permanent equity based on the terms of the Merger. The issuance of
Note 8 STOCK-BASED COMPENSATION
Equity Incentive Plan
The Company maintains the 2024 Equity Incentive Plan (the “Plan”),
under which the Company may grant stock options, restricted stock units, and other equity awards to employees, directors, and consultants.
As of December 31, 2025,
Stock Option Activity
| Number of Options | Weighted Average Exercise Price | Weighted-Average Remaining Contractual Term (In Years) | ||||||||||
| Issued and outstanding, December 31, 2024 | $ | |||||||||||
| Granted | $ | |||||||||||
| Exercised | - | |||||||||||
| Forfeited/Expired | - | |||||||||||
| Issued and outstanding, December 31, 2025 | $ | |||||||||||
| Exercisable at December 31, 2025 | $ | |||||||||||
Stock-Based Compensation Expense
| Year Ended | Year Ended | |||||||
| 12/31/2025 | 12/31/2024 | |||||||
| Research and development | $ | $ | ||||||
| General and administrative | ||||||||
| Total stock-based compensation | $ | $ | ||||||
F-17
As of December 31, 2025, total unrecognized compensation cost related to
unvested awards was $
Parent Awards to Subsidiary Employees
Certain equity awards of the Company have been granted to employees who
are now employees of Lokahi Therapeutics (“the BioBusiness”). Because there is no recharge arrangement (an agreement in which
the subsidiary reimburses the parent for the cost of stock-based awards granted to the subsidiary’s employees), between the Company and
the BioBusiness, the Company recognizes the stock-based compensation expense associated with these awards in its consolidated statement
of operations. In the standalone financial statements of Lokahi, the expense is offset by a corresponding capital contribution from the
Company. For the year end December 31, 2025, stock-based compensation of $
Note 9 WARRANTS
Outstanding Warrants
As of December 31, 2025, the following warrants were outstanding:
| Description | Shares | Exercise Price | Classification | |||||||
| Representative Warrants | ||||||||||
| Representative Warrants | ||||||||||
| Advisory Warrants | ||||||||||
| Total | ||||||||||
Reclassification from Liabilities to Equity
During the year ended December 31, 2025, the Company reclassified warrants
previously classified as liabilities to stockholders’ equity. Prior to the reclassification, the warrants were measured at fair value
at each reporting date in accordance with ASC 815-40, with changes in fair value recognized within “Change in fair value of warrant
liabilities” in the consolidated statement of operations. The reclassification occurred on August 5, 2025, as a result of official
issuance. On the date of reclassification, the warrants had a fair value of $
Fair Value Changes Prior to Reclassification
For the year ended December 31, 2025, the Company recognized a loss of
$
Note 10 Net Loss Per Share
The Company’s basic net loss per share is calculated by dividing net loss by the weighted-average number of shares of common stock outstanding for the period.
Diluted net loss per share is computed by giving effect to all potential shares of common stock, to the extent dilutive, including shares underlying the Series A convertible preferred shares, senior secured convertible notes, stock options, and stock warrants. Potential shares of common stock are excluded from the computation of diluted net loss per share if their effect would have been anti-dilutive for the periods presented or if the issuance of shares is contingent upon events that did not occur by the end of the period.
F-18
The following table encapsulates all potential shares of common stock that were excluded from the computation of weighted-average diluted shares.
| Common Share Equivalents | ||||
| Series A convertible preferred shares ( | ||||
| Stock options | ||||
| Warrants | ||||
| PIPE convertible note | ||||
| Total anti-dilutive shares excluded | ||||
Note 11 LEASES
Operating Lease
On December 12, 2025, the Company entered into an operating lease for office
space located in San Diego California, United States. The lease has a term of
As of December 31, 2025, the Company had made only the initial signing
payment of $
Balance Sheet Classification
Operating Lease
| December 31, 2025 | ||||
| Right-of-use asset, net | $ | |||
| Lease liability — current | $ | |||
| Lease liability — non-current | ||||
| Total lease liability | $ | |||
Lease Cost
| Year Ended 12/31/2025 | ||||
| Operating lease cost | $ | |||
| Short-term lease cost | ||||
| Total lease cost | $ | |||
Future Minimum Lease Payments
| Year Ending December 31, 2025 | Amount | |||
| 2026 | $ | |||
| 2027 | ||||
| 2028 | ||||
| Thereafter | ||||
| Total undiscounted lease payments | ||||
| Less: imputed interest | ( | ) | ||
| Present value of lease liabilities | $ | |||
F-19
Supplemental Information
| Year Ended 12/31/2025 | ||||
| Cash paid for amounts included in lease liabilities | $ | |||
| Weighted-average remaining lease term (years) | ||||
| Weighted-average discount rate | % | |||
Note 12 RELATED PARTY AND INTERCOMPANY TRANSACTIONS
Related Party Transactions
On March 21, 2025, the Company received $
During the year, an officer of the Company advanced
the Company $
Note 13 INCOME TAXES
Income Tax Expense (Benefit)
The Company recorded income tax expense (benefit) of for the year ended December 31, 2025.
Effective Tax Rate Reconciliation
The Company adopted Accounting Standards Update (ASU) 2023-09, “Improvements to Income Tax Disclosures,” on a retrospective basis within its annual reporting for the year ended December 31, 2025. The adoption of ASU 2023-09 resulted in enhanced disclosures related to the effective tax-rate reconciliation, including additional disaggregation requirements prescribed by the standards.
During 2025, the Company elected accelerated amortization under the transition
provisions of the One Big Beautiful Bill Act for previously capitalized domestic research and experimental expenditures.
| For the years ended December 31, | ||||||||||||||||
| 2025 | 2024 | |||||||||||||||
| U.S. Federal statutory tax rate | ( | ) | % | $ | ( | ) | % | |||||||||
| State and local income tax, net of federal income tax effect | ||||||||||||||||
| New Jersey | ( | ) | % | ( | ) | % | ||||||||||
| Valuation allowance | - | % | - | % | ||||||||||||
| Changes in valuation allowances | - | % | - | % | ||||||||||||
| Nontaxable or nondeductible items | ||||||||||||||||
| Accretion expense | - | % | - | % | ||||||||||||
| Other | % | % | ||||||||||||||
| Other Adjustments | ||||||||||||||||
| Intangible true-up | ( | ) | % | ( | ) | % | ||||||||||
| Income tax | $ | % | $ | % | ||||||||||||
F-20
Deferred Tax Assets and Liabilities
| For the years ended December 31, | ||||||||
| 2025 | 2024 | |||||||
| Net operating loss carry forwards | $ | $ | ||||||
| Stock based compensation | ||||||||
| Accruals | ( | ) | ||||||
| Capitalized research and development | ||||||||
| Intangible assets | ( | ) | ||||||
| Fixed Assets | ( | ) | ||||||
| Right of use assets | ( | ) | ||||||
| Change in fair value of digital assets | ( | ) | ||||||
| Change in fair value of warrant liabilities | ( | ) | ||||||
| Change in fair value of derivatives | ( | ) | ||||||
| Total deferred tax assets | ||||||||
| Valuation allowance | ( | ) | ( | ) | ||||
| Net deferred tax assets | $ | $ | ||||||
The Company has cumulative federal net operating losses of $
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Deferred tax assets consist primarily of the tax effect of NOL carry-forwards. The Company has provided a full valuation allowance on the deferred tax assets because of the uncertainty regarding its realizability.
The Company’s policy is to record interest and penalties associated with unrecognized tax benefits as additional income taxes in the statement of operations. As of December 31, 2025, the Company had no unrecognized tax benefits. There were changes in the Company’s unrecognized tax benefits during the years ended December 31, 2025 and 2024. The Company did not recognize any interest or penalties during the 2025 fiscal year related to unrecognized tax benefits.
F-21
Note 14 COMMITMENTS AND CONTINGENCIES
License Agreement
On August 2, 2021, the Company entered into a
business agreement with Apimeds Korea. Under the agreement, the Company received the right to continue any clinical trial and acquire
the permits and approval necessary from the U.S. Food and Drug Administration. The Company will pay Apimeds Korea a royalty of
Legal Proceedings
From time to time, the Company may be involved in legal proceedings arising in the ordinary course of business. As of December 31, 2025, the Company was not a party to any legal proceedings that management believes would have a material adverse effect on the Company’s financial position, results of operations, or cash flows.
Future Commitments
During the year ended December 31, 2025, the Company entered into an agreement
to accumulate a prepaid balance with its respective Clinical Research Organization, Prevail InfoWorks Inc, pertaining to future clinical
trial execution. The total remaining obligation associated with this agreement is $
During the year ended December 31, 2025 the Company entered into an agreement with Piramal Pharma Solutions, Inc. to manufacture clinical trial material for its lead Biopharmaceutical asset, Apitox.
The Company holds customary employment agreements
with its key executives. These employment agreements provide for compensation in the form of salary, employee benefits, stock compensation,
discretionary bonuses. The contract in place for the President of Lokahi Therapeutics (“The BioBusiness”) includes a severance
package equivalent to twenty four (24) months of salary and benefits, or $
Indemnification Agreements
The Company has entered into indemnification agreements with its directors and officers. Under these agreements, the Company may be required to indemnify its directors and officers against certain liabilities that may arise by reason of their status or service. The Company has not incurred material costs related to these indemnification provisions and has not accrued any liabilities related to such obligations as of December 31, 2025.
Note 14 SUBSEQUENT EVENTS
The Company has evaluated subsequent events the date on which the consolidated financial statements were available to be issued.
| ● | Pursuant to the securities purchase agreement outlined in note 6, the company expects to continue funding operations through the execution of additional convertible notes in accordance with the maximum aggregate principal to fund operations of the parent, and its respective subsidiaries. |
| ● | On March 30, 2026, the BioBusiness issued a $ |
|
| ● | On February 5, 2026, MindWave
issued a $ | |
| ● | On April 24, 2026, the Company and its respective subsidiaries
entered into a Settlement Agreement which resolves all outstanding disputes among related parties. Pursuant to the Settlement Agreement,
the Company plans to issue |
F-22