10-K: Annual report [Section 13 and 15(d), not S-K Item 405]
Published on April 15, 2025
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
For the fiscal year ended
For the transition period from to
Commission File Number:
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) |
|
| (Address of principal executive offices) | (Zip Code) |
(Registrant’s telephone number, including area code)
Not applicable
(Former name, former address and former fiscal year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a
well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not
required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant
(1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months
(or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements
for the past 90 days.
Indicate by check mark whether the registrant
has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (Section 232.405
of this chapter) during the preceding 12 months (or such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| ☒ | Smaller reporting company | ||
| Emerging growth company |
If an emerging growth company, indicate by check
mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting
standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant
has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial
reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or
issued its audit report.
If securities are registered pursuant to Section
12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction
of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant
is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
As of June 28, 2024, the last business day of
the registrant’s last completed second quarter, there was
As of April 15, 2025, there were
TABLE OF CONTENTS
i
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
Various statements in this Annual Report on Form 10-K (the “Annual Report”) of Apimeds Pharmaceuticals US, Inc. are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements involve substantial risks and uncertainties. All statements, other than statements of historical facts, included in this report, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management are forward-looking statements. These statements are subject to risks and uncertainties (some of which are beyond our control) and are based on information currently available to our management. Words such as “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “plan,” “contemplates,” “predict,” “project,” “target,” “likely,” “potential,” “continue,” “ongoing,” “will,” “would,” “should,” “could,” or the negative of these terms and similar expressions or words, identify forward-looking statements. The events and circumstances reflected in our forward-looking statements may not occur and actual results could differ materially from those projected in our forward-looking statements. Such forward-looking statements are based on current expectations and involve inherent risks and uncertainties, including risks and uncertainties that could delay, divert or change these expectations, and could cause actual results to differ materially from those projected in these forward-looking statements. These risks and uncertainties include, but are not limited to, those factors described under Part I, Item 1A: “Risk Factors.” Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements.
This Annual Report contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this report is generally reliable, such information is inherently imprecise and subject to change.
All written and oral forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We caution investors not to rely on the forward-looking statements we make or that are made on our behalf as predictions of future events. We undertake no obligation and specifically decline any obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws.
We encourage you to read the management’s discussion and analysis of our financial condition and results of operations and our financial statements contained in this Annual Report. There can be no assurance that we will in fact achieve the actual results or developments we anticipate or, even if we do substantially realize them, that they will have the expected consequences to, or effects on, us. Therefore, we can give no assurances that we will achieve the outcomes stated in those forward-looking statements, projections and estimates.
ii
PART I
Item 1. Business
We are a clinical stage biopharmaceutical company in the process of developing Apitox, an intradermally administered bee venom-based toxin. Our focus is primarily on developing innovative therapies that address inflammation and pain management symptoms associated with knee OA and, to a lesser extent, MS. Apitox is currently marketed and sold by Apimeds Inc. (“Apimeds Korea”) in South Korea as “Apitoxin” for the treatment of OA. Apimeds US is not associated with the market, sale and revenues generated from Apitoxin in South Korea, and Apitoxin has not yet been approved by the FDA for any indication.
Apitox is a purified, pharmaceutical grade venom (bee venom), of the Apis mellifera, or western honeybee, which is classified by the FDA as an active pharmaceutical ingredient (“API”). Bee venom has been used in Asia and Europe to treat pain for hundreds of years. While not FDA approved in a controlled, prescription based biologic environment for defined indications, the use of bee venom has been FDA approved as a “under the skin injection” to reduce the allergic reactions to bee stings. Apimeds Korea has developed a proprietary method and process for turning extracted bee venom into a lyophilized powder for reconstitution prior to intradermal dose injections, which they sell in South Korea as Apitoxin. We intend to use a similar process with respect to Apitox, pursuant to the Business Agreement, which gives us a license to utilize all prior clinical development data associated with Apitoxin. The advancement of extracted bee venom for treatment of inflammatory conditions, including but not limited to knee OA and MS is speculative but based on direction provided by prior clinical data.
Apimeds Korea successfully completed Phase I, Phase II, and Phase III trials in OA in 2003, at which point Apitoxin was approved by the Korean Ministry of Food and Drug Safety (“MFDA”) to treat pain and mobility in patients with OA. Since 2003, a post-marketing/approval safety study in South Korea followed 3,194 patients from 2003 through 2009, with no serious adverse events. The purpose of a Phase I trial is to test to determine whether a new treatment is safe and look for the best way to give the treatment. Phase II trials test to determine whether a condition or disease responds to the new treatment. Phase III trials test to determine whether a new treatment is better than a standard treatment.
In 2013, the first of two required U.S. Phase III clinical trials was authorized to enroll patients to study the use of Apitoxin to study the same indication as approved in South Korea in 2023 — treatment of pain and lack of mobility in patients with OA (the “Apimeds Korea Phase III OA Trial”). The Apimeds Korea Phase III OA Trial (330 patients) was completed in 2018, and displayed no serious adverse events.
Based on the results from the Apimeds Korea Phase III OA Trial, which demonstrated therapeutic (statistical and clinically significant improvements in all outcome measures of pain, physical function, and disease assessment) effect compared to the placebo group, but in combination with prior development by Apimeds Korea, did not meet the FDA’s standards for approval, as the study population was too small and the methods for handling missing data were inadequate, resulting in a study that did not demonstrate a significant treatment effect. We will be pursuing a second Phase III trial to meet agreed upon FDA standards. Based on results from the Apimeds Korea Phase III OA Trial, we have evaluated the most appropriate population, defined as advanced knee OA patients, which will range from defined grade 2, 3 and 4 within this treatment group, to continue to progress our own Phase III trial. Pursuant to our previous correspondence with the FDA, we have designed and will implement our Phase III trial to best address our patient population, appropriate dosing, and the most effective way to evaluate Apitox in meeting the patient population’s needs.
We believe the progress we are making in clinical trials provides us support in our belief in the potential of Apitox to be an innovative therapy. We aim to treat the inflammation and pain management symptoms associated with knee OA and to help manage the devastating symptoms of this disease. In the future, we also aim to leverage our research in knee OA to investigate how Apitox may be used to treat similar symptoms associated with MS.
Treatment of OA
OA is typically treated with painkillers known as non-steroidal anti-inflammatory drugs (NSAIDs). These medications have an anti-inflammatory and pain-relieving effect. These medications include ibuprofen (Motrin, Advil) naproxen (Aleve) and diclofenac (Voltaren and others). All of these medications work by blocking enzymes that cause pain and swelling. The problem is that some of those enzymes also help blood to clot and protect the lining of your stomach. Without them, you can bruise easily, develop ulcers and may even bleed in your intestines. NSAIDs also increase your chance of heart attack, stroke and heart failure. The risk increases the longer you use them and the more you take. We believe Apitox could be a successful alternative to NSAIDs in the treatment of the inflammation and pain management symptoms associated with OA without the harmful side effects.
1
According to MedicalNewsToday, OA is the most common form of arthritis, affecting around 500 million people worldwide, or around 7% of the global population. Currently, in the United States, over 32 million people suffer from OA. As the 15th highest cause of years lived with disability (YLDs) worldwide, the burden OA poses to individuals is substantial, characterized by pain, activity limitations, and reduced quality of life. The economic impact of OA, which includes direct and indirect (time) costs, is also substantial, ranging from 1 to 2.5% of gross national product (GNP) in countries with established market economies, like the United States. Though trends in OA prevalence vary by geography, the prevalence of OA is projected to rise in regions with established market economies such as North America and Europe, where populations are aging and the prevalence of obesity is rising.
While OA can occur in any joint, it occurs most frequently in the knee, which, according to ScienceDirect, currently accounts for 365 million cases worldwide and 61% of YLDs lost due to OA, followed by the hand.
Our current efforts are focused on the development of Apitox in the United States for the treatment of inflammation and pain management relating to OA in the knee.
Treatment of MS
Additionally, we believe the previous clinical trial success of Apimeds Korea with respect to the use of Apitoxin to treat symptoms associated with knee OA, and pending the success of our anticipated Phase III trial in knee OA, we will be in a position to further explore the use of Apitox as a potential treatment for the symptoms of MS. MS is a chronic disease of the central nervous system. It is an autoimmune condition that is characterized by the body’s own immune cells (macrophages and lymphocytes) attacking the myelin that coats nerve cells, which can lead to inflammation throughout the central nervous system. MS is an unpredictable disease that affects people differently. Some people with MS may have only mild symptoms. Others may lose their ability to see clearly, write, speak, or walk when communication between the brain and other parts of the body becomes disrupted.
MS is the most common progressive neurologic disease of young adults worldwide. A study funded by the National MS Society estimates that nearly one million individuals are currently affected by this disease in the United States. The total economic burden of MS in the United States is estimated to be $85.4 billion, with $63.3 billion in direct medical costs and $22.1 billion in indirect and nonmedical costs. MS typically affects patients at a young age, resulting in a greater loss of productivity and quality of life.
Beta interferon drugs are among the most common medications used to treat MS. Interferons are signaling molecules that regulate immune cells. Potential side effects of these drugs include flu-like symptoms (which usually fade with continued therapy), depression, or elevation of liver enzymes.
Pain from MS can be felt in different parts of the body. Trigeminal neuralgia (facial pain) is treated with anticonvulsant or antispasmodic drugs, or less commonly, painkillers. Central pain, a syndrome caused by damage to the brain and/or spinal cord, can be treated with gabapentin and nortriptyline. Treatments for chronic back or other musculoskeletal pain may include heat, massage, ultrasound, and physical therapy.
OA and the Current Standard of Care
OA is a degenerative joint disease in which the tissues in the joint break down over time. It is the most common type of arthritis and is more common in older people. People with osteoarthritis usually have joint pain and, after rest or inactivity, stiffness for a short period of time.
There are four stages of OA: (1) Minor — minor wear-and-tear in the joints and little to no pain in the affected area, (2) Mild — more noticeable bone spurs, the affected area feels stiff after sedentary periods and patients may need a brace, (3) Moderate — cartilage in the affected area begins to erode, the joint becomes inflamed and causes discomfort during normal activities, and (4) Severe — the patient is in a lot of pain, the cartilage is almost completely gone leading to an inflammatory response from the joint, and overgrowth of bony spurs may cause severe pain.
2
With the progression of OA of the knee, there is obvious joint inflammation which causes frequent pain when walking, running, squatting, extending or kneeling. Along with joint stiffness after sitting for long or when waking up in the morning, there may be popping or snapping sounds when walking.
The data from the Apimeds Korea Phase III OA Trial suggest that Apitox would have the most potential in treating OA in stages 3 and 4.
MS and the Current Standard of Care
MS is increasingly recognized as a neurodegenerative disease triggered by an inflammatory attack of the central nervous system. There is no cure for multiple sclerosis. Treatment typically focuses on speeding recovery from attacks, reducing new radiographic and clinical relapses, slowing the progression of the disease, and managing MS symptoms.
MS is unpredictable and can vary substantially from person to person. MS is divided into four types: clinically isolated syndrome (CIS), relapsing-remitting MS (RRMS), secondary progressive MS (SPMS) and primary progressive MS (PPMS).
CIS refers to a first episode of neurologic symptoms caused by inflammation and demyelination in the central nervous system.
RRMS, the most common disease course, shows clearly defined attacks of new or increasing neurologic symptoms. These attacks are also called relapses or exacerbations. They are followed by periods of partial or complete recovery, or remission. In remissions, all symptoms may disappear or some symptoms may continue and become permanent. However, during those periods, the disease does not seem to progress.
SPMS follows the initial relapsing-remitting course. Some people diagnosed with RRMS eventually go on to have a secondary progressive course, in which neurologic function worsens progressively or disability accumulates over time.
With PPMS, neurologic function worsens or disability accumulates as soon as symptoms appear, without early relapses or remissions. PPMS can be further characterized as either active (with an occasional relapse and/or evidence of new MRI activity over a specified period of time) or not active, as well as with progression (evidence of disability accrual over time, with or without relapse or new MRI activity) or without progression.
Patients with MS tend to be more educated about their disease and better organized than patients with other diseases, resulting in patients that are aggressive in their approach to treatment. This is due to MS impacting otherwise healthy people in the prime of their lives.
MS treatment has undergone significant evolution in the last ten years with the development and approval of certain new drugs, including several oral agents such as Ocrevus, in the United States. These new agents not only give patients additional treatment options, but also have improved the efficacy and safety of treatment for MS overall. In general, these drugs are “disease modifying agents,” intended to slow down the immune mediated damage to the myelin sheaths that underlie symptoms in MS. However, they often do not adequately address the symptoms that MS patients experience such as walking problems, bladder control, dizziness, and especially pain. A 2022 study estimated that the average cost of treatment for patients with MS is approximately $88,000 annually. The out-of-pocket expense for patients can be significantly reduced through certain insurance plans. However, we believe there is the ability for Apitox to be positioned as an important and cost-effective therapy.
We believe the data from the Apimeds Korea Phase III OA Trial suggest that Apitox may have the potential as an adjunctive therapy for all four types of MS. We intend to Apitox as a potential adjunctive therapy through non-registered corporate sponsorship studies to begin determining the appropriate MS patient populations.
Market Opportunity
We believe there is a significant market opportunity in the United States for Apitox in the treatment of certain symptoms of knee OA and eventually MS. According to Precedence Research the osteoarthritis therapeutics market size accounted for $8.28 billion in 2022 and it is expected to hit around $20.24 billion by 2032, expanding at a CAGR of 9.4% from 2023 to 2032. Although OA can damage any joint, the disorder most commonly affects joints in your hands, knees, hips and spine. OA symptoms can usually be managed, although the damage to joints can’t be reversed. Apitox has certain anti-inflammatory properties, which we believe give it significant potential to help treat the symptoms of certain chronic diseases that involve difficult to control pain and inflammation.
3
According to Pharmaceutical Technology the MS market size in the United States accounted for $10.73 billion in 2022 and is expected to hit $24.4 billion by 2030, expanding at a CAGR of 10.32%. Starting in the first quarter of 2025, we intend to begin the early prosecution of appropriate MS patient populations through non-registered corporate sponsorship studies. Subject to FDA approval, our development of Apitox in the United States will in the near term, have two distinct focuses (i) the treatment of the certain symptoms of knee OA and (ii) the quality of life issues surrounding knee OA, such as pain and lack of mobility.
Living with a chronic disease is challenging, as it interferes with physical, mental, and social functions and thus greatly affects a person’s quality of life. Indeed, chronically ill patients are facing major struggles such as higher expenditures, social isolation and loneliness, disabilities, fatigue, pain/discomfort, feelings of distress, anger, hopelessness, frustration, anxiety, and depression. There is the general assumption that symptom reduction increases a patient’s quality of life. Our approach with Apitox centers around this concept — effectively treating certain symptoms of the patient’s disease, thus improving their overall quality of life. Bee venom has been shown to have anti-inflammatory effects. At low doses, bee venom can suppress inflammatory cytokines such as interleukin-6 (IL-6), IL-8, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α). A decrease in the signaling pathways responsible for the activation of inflammatory cytokines, such as nuclear factor-kappa B (NF-κB), extracellular signal-regulated kinases (ERK1/2) and protein kinase Akt, and porphyromonas gingivalis lipopolysaccharide (PgLPS)-treated human keratinocytes has been associated with treatments involving bee venom. We believe the driver of pain in the highest category of OA is correlated to the key inflammatory elements treated by bee venom, meaning the evaluation of our Phase III data may lead to a small indication for narcotic use reduction in the treatment of stage 4 OA.
Our Product Candidate
Apitox is purified honeybee (Apis mellifera) venom manufactured as a lyophilized powder for reconstitution in 0.5% preservative-free lidocaine (lmg/mg) prior to intradermal dose injections that are administered up to 1,500 micrograms per weekly visit. The biologically active components include melittin (40-50%), apamin (2-3%), mast cell degranulating (“MCD”) peptide (Peptide 401,2-3%), phospholipase A2 (10-15%), hyaluronidase (1.5-2%) and other components in small amounts, including dopamine and norepinephrine. According to a publication entitled “Pharmacological effects and mechanisms of bee venom and its main components: Recent progress and perspective” by Shi et al., certain components of honeybee venom have been found to have both anti-inflammatory and analgesic effects. The anti-inflammatory and analgesic effects are attributed to the presence of Peptide 401, adolapin and other components that inhibit prostaglandin synthesis. The hormone-stimulating effects are attributed to the presence of melittin, cardiopep and other components that stimulate the pituitary-adrenal axis to produce cortisol. Results from an animal study entitled “Effect of bee venom and melittin on plasma cortisol in the unanesthetized monkey” published by Vick et al., indicate that melittin appears to stimulate the production of cortisol from the adrenal gland. The immune-modulating effects, especially as it pertains to MS, are suggested to be mediated by CD4+CD2S+Foxp3+ regulatory T cells (Tregs) that are influenced by phospholipase A2. While the exact mechanism of action of Apitox is not fully understood, research such as the publication entitled “Therapeutic Use of Bee Venom and Potential Applications in Veterinary Medicine” by Bava et al., suggests that certain components in Apitox may ameliorate immune-inflammatory responses associated with MS. Such studies suggested that treatments with melittin prevent inflammatory cytokine expression and produces anti-inflammatory effects. The proposed indication for Apitox is to provide add-on therapy for the signs and symptoms of MS in patients whose condition is relapsing-remitting (RRMS), primary-progressive (PPMS) or secondary progressive (SPMS).
4
Clinical Development History
Founded in 1989, Apimeds Korea pursued a traditional drug development process in South Korea for Apis mellifera, the bee venom API for Apitoxin. Apimeds Korea completed a formal preclinical study to validate dosing and safety for human administration with a focus on antigenicity and toxicology in 1993.
A Phase I trial was completed in 1994, studying the toxicity and safety of Apitoxin in 20 healthy subjects. The purpose of the Phase I trial was to determine if therapeutic doses of Apitoxin was safe and to identify possible side-effects, if any. Injections of Apitoxin were given two to three times a week, for a total of 12 sessions spanning over four to six weeks. Laboratory and physical examination of the subjects included (i) serum cortisol levels (to see if Apitoxin stimulated the release of cortisol), (ii) serum ionized calcium level (to determine if Apitoxin decreased the serum calcium level), (iii) urinalysis, (iv) hematology and blood chemistry, and (v) vital signs. The Phase I trial demonstrated that there were no significant changes pre- and post-testing of the serum cortisol levels, serum ionized calcium levels, hematology, blood chemistry, urinalysis, and vital signs after the subjects were injected with Apitoxin according to the protocol. There were no significant physiological changes in the clinical evaluations of the subjects and localized itching was the most frequent side effect and was managed with ice packs or external anti-itching gels. No severe side effects or aftereffects were observed. The Phase I trial indicated that Apitox is safe for humans when applied in therapeutic doses.
The Phase I trial was followed by a Phase II trial in 101 subjects to determine the efficacy of Apitoxin at various dose levels. This was a randomized active-controlled clinical trial with three groups receiving the study drug at various dose levels and one group receiving the control drug (nabumetone) for a six-week period. Patients received twice weekly injections of Apitox intradermally at dosages titrated to a maximum of 0.7 mg (Group A), 1.5 mg (Group B), and 2.0 mg (Group C) for a period of six weeks. Control group patients (Group D) received 1,000 mg of nabumetone orally each day for the same six-week period. There were 25, 26, 25 and 25 patients assigned to Groups A, B, C and D, respectively. Efficacy of treatment was evaluated by the physician investigators using a 4-point Likert-like symptom severity rating scale developed by the authors to assess Pain, Disability and Physical Signs. A similar 5-point scale was used for patient self-evaluation. Safety of the Apitoxin injection was evaluated by patient reaction, hematologic examination, and laboratory chemistry analysis of blood and urine. Efficacy data was reported for the 81 patients who completed the study. While there were no significant differences in symptom severity scores among the four groups at baseline, symptom scores were significantly better in the bee venom injection groups than in the control group at six weeks and 10 weeks after the start of treatment (p<0.01). A treatment was considered effective if there was a 20% improvement from baseline in symptom scores after 6 weeks of treatment. Based on this definition, therapy demonstrated overall efficacy in 70.0% of patients in Group A, 85.7% in Group B, 90.0% in Group C, and 61.9% in Group D (drug control). Overall efficacy was significantly greater in treatment Groups B and C combined than in the nabumetone-treated control group D (p<0.0177). Importantly, efficacy of treatment among all patients treated with Apitoxin injection was greater than among nabumetone-treated patients for each category assessed: Pain: 85.2% versus 76.2%; Disability: 77.0% versus 71.4%; and Physical Signs: 62.3% vs. 23.8%. It is also noteworthy that, unlike the drug control group, the Apitoxin injection groups continued to demonstrate improved symptom scores at four weeks after the last treatment (10 weeks). There were no significant changes in vital signs or results of laboratory examinations of any patient in this clinical trial. Localized itching was experienced by all patients who received Apitox injections. Itching at the injection site generally lasted for two to three weeks; several patients had this reaction for a longer period. This Phase II study showed that Apitoxin was significantly more effective than the control drug, nabumetone, in the treatment of knee and spinal osteoarthritis patients. It clearly showed that improvement in pain, disability and physical signs was greater in the bee venom injection groups than in the nabumetone control group. No significant side effects developed at the therapeutic doses studied. However, research should be continued to minimize itching and pain at bee venom injection sites, and possible allergic reaction should always be considered with treatment at high doses.
5
In 2002, a formal Phase III double-blind, placebo-controlled trial was completed with 407 subjects (311 of which obeyed the trial protocol and completed the clinical study). The purpose of the Phase III trial was conducted to verify the efficacy and safety of the medicine resulting from the prior Phase I and Phase II trials. The therapeutic course treatment included a total of 12 injections over a period of 6 weeks. Final evaluations were completed in the 8th week, following two weeks of no injections. During the trial period, laboratory tests were carried out three times (before injection, in the second week, in the sixth week), and the efficacy evaluation was performed four times (before injection, in the second week, in the sixth week, and in the eighth week). Safety of the Apitoxin injection was evaluated by, hematologic examination, measurement of cortisol and calcium levels, and laboratory chemistry analysis of blood and urine. The primary efficacy variable for the trial was the ratio of the subjects who showed more than 20% improvement in the total points of test items for efficacy evaluation 6 weeks after injection, compared with the total points before injection of the medicine (the “improvement rate”). Data obtained from subjects of the clinical test were analyzed by two methods, ITT (Intention to Treat) analysis and PP (Per Protocol) Among 310 subjects who participated in the efficacy evaluation, 153 and 157 patients belonged to the Apitoxin group and the nabumetone group, respectively. For the Apitoxin group, the ratio of the subjects who showed more than 20% improvement in the total points was 48.70% (75/154 subjects, 95% confidence interval (“CI”): 40.8~56.6%), while for the nabumetone group, it was 46.15% (72/156 subjects, 95% CI: 38.3~54.0%), indicating that the improvement rate in the Apitoxin group was greater than in the nabumetone group; however, there was no statistical significance. (p=0.6533). Among a total of 407 subjects (Apitoxin group: 204; Nabumetone group: 203), 38.24% (78/204) of the Apitoxin group showed more than 20% improvement during the 6th week of injection, while 38.42% of the Nabumetone group improved by more than 20%, indicating that the two groups showed similar improvement rate (p=0.9688). The second efficacy variable was the improvement rate during the 8th week (2 weeks after the completion of the final injection). According to results from comparing the total points of efficacy evaluation items during the second week after completion of injection (during the 8th week after injection) with the total points before injection, 58.44% (90/154) of the Apitoxin group showed a higher improvement rate than during the 6th week (48.70%), while 42.95% (67/156) of the Nabumetone group showed lower improvement rate than during the 6th week (46.15%). There was statistical difference in total point of efficacy evaluation items between the two groups (p=0.0064). These results suggest that even after treatment stops, the efficacy of Apitoxin continues. With respect to safety, among a total of 407 subjects who participated in the safety evaluation, 69 (33.82%) of the Apitoxin group showed an adverse event, while 59 (29.06%) of the Nabumetone indicated adverse event. These results indicate that the Apitoxin group had an elevated adverse event rate than the Nabumetone group, but there was no statistically significant difference between the two groups (p=0.3526).
In May 2003, MFDA granted approval for the use of Apitoxin in the treatment of pain and mobility in patients with OA. A post-marketing/approval safety study in South Korea followed 3,194 patients from 2003 through 2009, with no serious adverse events or negative safety signals.
In 2013, preliminary Phase III clinical trials were authorized to enroll patients by the FDA to study the same indication approved in South Korea — treatment of pain and lack of mobility in patients with OA. The results of the preliminary Phase III clinical trial indicated statistical and clinically significant improvements in all outcome measures of pain, physical function, and disease assessment in the study group. The study group included 330 patients with diagnosed osteoarthritis of the knee. The subjects were evaluated for relief of pain using Western Ontario and McMaster Osteoarthritis Index (WOMAC) and physician and patient global assessments. The primary efficacy measure was relief of pain and inflammation over a 12-week treatment period after randomization into the trial. The secondary efficacy measure was improvement of mobility. Treatment effect will be compared in a 2-1 Apitox vs active control. Compared with the placebo group (histamine), subjects in the Apitox group who received a maximum dose (1500 micrograms) at each weekly visit over 12 weeks showed a significantly more improvement in all outcome measures (WOMAC pain, WOMAC physical function, visual analog scale (“VAS”) pain, patient and physician global assessments of OA). Further, post hoc analyses showed that a statistically significant greater percentage of Apitox-treated subjects had at least a 40% and 60% reduction in WOMAC pain as compared to placebo-treated subjects. Sensitivity analyses confirmed the validity of the statistical methods and population definitions. The improvements in pain endpoints were highly significant for both the modified intention to treat and per protocol populations and the improvement was sustained during the four weeks following Apitox treatment.
Except for an expected higher incidence of injection site reactions (<5%) in the Apitox group, the overall safety profiles were comparable between the treatment groups. A serious adverse event of the anaphylactic reaction occurred in an Apitox-treated subject because of a quick injection rate. However, the subject was treated, and the event was resolved within one day. The incidence of adverse events overall was similar between the Apitox and Placebo groups (49.0% and 46.3%, respectively), and there were no clinically meaningful changes, within and between groups, in laboratory parameters, vital signs, physical examination, or electrocardiogram results.
6
During Apimeds Korea meetings with the FDA, the FDA highlighted concerns regarding the opioid crisis. As Apitoxin has been previously approved in South Korea, we believe Apitox could be a viable treatment option within the United States after additional clinical investigation, including our anticipated Phase III trial. Initially, Apimeds Korea elected not to pursue the OA indication in the United States based on its evaluation of potential market adoption and the existing competitive environment for OA. Based on results from the Apimeds Korea Phase III OA Trial and correspondence with the FDA, we believe we are now in a position to continue to advance our Phase III trial for knee OA.
We intend to conduct an additional Phase III trial in knee OA. Based on our previous correspondence with the FDA, we have started to design and will implement our Phase III trial to best address our patient population of patients with grade 2, 3 and 4 knee OA, appropriate dosing, and the most effective way to evaluate Apitox in meeting a patient’s needs. This trial will be an update to the plan of execution based on review of data, discussions with former principal investigators from Apimeds Korea. Upon successful completion and FDA clearance of our Phase III trial in knee OA, we will be positioned to submit a BLA.
We intend that the purpose of this trial will be to evaluate the effectiveness of Apitox in the treatment of grade 2, 3 and 4 OA of the knee. The trial will be designed with a specific focus on the identified subgroup from which we see the highest degree of benefit.
The following table summarizes the preliminary clinical trial activity by Apimeds Korea with respect to Apitoxin:
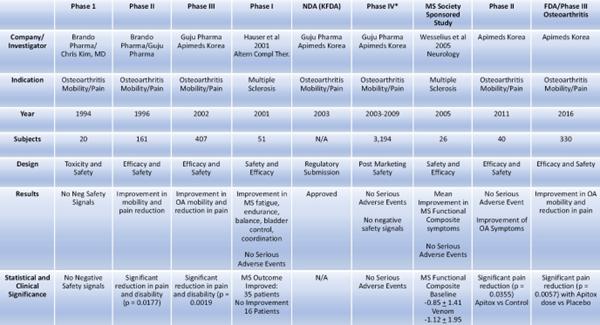
Preliminary Clinical Data in MS Patients
The United States data from the literature on bee venom studies in MS patients, Table A (Hauser et al. 2001) below, showed clinically significant improvements in disability symptoms following treatment.
In Table A, results were categorized into the following groups: dramatic disability improvement (>12 points on the Related Observable Symptom Scale (“ROSS”), good improvement (7-12 points on ROSS), minimal improvement (<7 points on ROSS), no improvement (<2 points on ROSS), and negative (any total negative response on ROSS). Descriptive analysis of the ROSS clinical outcomes showed that more than 68% of MS patients showed some kind of positive improvement in disability (dramatic, good or minimal) and 58% demonstrated a marked improvement (dramatic or good).
Table A. Summary of Patient Disability Improvement to Bee Venom Treatment Using ROSS
| N | % of Participants |
Follow-up Survey (% improvement) |
Related Observable Symptoms Scale (points improvement) |
||||||
| Dramatic | 15 | 29.4 | % | >30%, or | >12 points | ||||
| Good | 15 | 29.4 | % | 10 – 29%, or | 7 – 12 points | ||||
| Minimal | 5 | 9.8 | % | <10%, or | <7 points | ||||
| None | 15 | 29.4 | % | <2%, or | <2 points | ||||
| Negative | 1 | 2.0 | % | Any total negative response | Any total negative response | ||||
7
After 1 year of bee-venom injections, 68.6 percent of participants showed improvement. N = number of participants.
Apimeds Korea used data from its first Phase III clinical trial for OA and peer reviewed publications, including those referenced in Table A above and formal Phase I (the “Castro Phase I Trial”) and Phase II (the “Wesselius Phase II Trial”) publications specific to MS, to support its submission in 2014 of its Investigational New Drug Application (“IND”) 122804 (A Phase III, Multi-Center, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study to Evaluate the Safety and Efficacy of Apitox Add-on Therapy for Improving Disability and Quality of Life in Patients with Multiple Sclerosis).
Castro Phase I Trial
The Castro Phase I Trial involved a total of nine bee venom nonallergic patients with progressive forms of MS, who were 21–55 years of age with no other illnesses. The subjects distributed across four groups (A, B, C, and D) and followed a structured 1-year immunization schedule. Hyperreactivity to bee venom was evaluated by questionnaire, physical examination, and a battery of hematologic, metabolic, and immunologic tests. Responses to therapy were evaluated by questionnaire, functional neurological tests, and changes in measurement of somatosensory-evoked potentials. While no serious adverse allergic reactions were observed in any of the subjects, four experienced worsening of neurological symptoms, requiring their discontinuation in the study. The observed negative effects could not be conclusively attributed to adverse reactions arising from the administered therapy. Of the remaining five subjects, three reported subjective amelioration of symptoms and two exhibited objective improvement. Despite suggesting safety in this preliminary study, the small sample size precluded definitive conclusions regarding the efficacy of the treatment for MS. Larger and more carefully conducted multicenter studies were required to establish efficacy.
Wesselius Phase II Trial
The Wesselius Phase II Trial involved a randomized crossover study of 26 patients diagnosed with relapsing-remitting or relapsing secondary progressive MS. Participants were assigned to 24 weeks of medically supervised bee sting therapy, or a control period of 24 weeks of no treatment. Live bees (up to a maximum of 20) were used to administer bee venom three times per week. The primary outcome was the cumulative number of new gadolinium-enhancing lesions on T1-weighted MRI of the brain. Secondary outcomes were lesion load on T2*-weighted MRI, relapse rate, disability (Expanded Disability Status Scale, Multiple Sclerosis Functional Composite, Guy’s Neurologic Disability Scale), fatigue (Abbreviated Fatigue Questionnaire, Fatigue Impact Scale), and health-related quality of life (Medical Outcomes Study 36-Item Short Form General Health Survey). The results of the Wesselous Phase II Trial indicated that during bee sting therapy, there was no significant reduction in the cumulative number of new gadolinium-enhancing lesions. The T2*-weighted lesion load further progressed, and there was no significant reduction in relapse rate. There was no improvement of disability, fatigue, and quality of life. Bee sting therapy was well tolerated, and there were no serious adverse events. In this trial, treatment with bee venom in patients with relapsing multiple sclerosis did not reduce disease activity, disability, or fatigue and did not improve quality of life measured using gadolinium-enhancing MRI.
From June 2014 to June 2018, Apimeds Korea corresponded with the FDA and there were no clinical holds at that time. Sponsorship of IND 122804 was transferred from Apimeds Korea to us in October 2020. On September 21, 2021, we responded to customary non-clinical hold comments from the FDA. In November 2021, we received a customary clinical hold from the FDA due to the retirement of the former principal investigator. We have subsequently updated the FDA with a new principal investigator via our Chief Medical Officer, Dr. Christopher Kim. In February 2023, the FDA removed the clinical hold and concluded it may be initiated. We have subsequently made the strategic decision to focus our efforts and capital on our Phase III trial in knee OA, and instead focus our MS efforts on the early prosecution of appropriate MS patient populations through non-registered corporate sponsorship studies.
Our Commercialization Strategy
We are dedicated to the effective implementation of regulatory, clinical and legal strategies to create value in Apitox. The effective execution of this strategy will provide us the opportunity to evaluate and potentially acquire other assets that fit within our space for development.
8
Manufacturing
We intend to continue to engage a third-party manufacturer, Piramal Pharma Solutions, in Lexington, Kentucky to support our Phase III trial and, if Apitox is approved by the FDA, commercial manufacturing. This manufacturer has dedicated experience in development and technology transfer of sterile dose formulations, including liquid and lyophilized formulations.
Research and Development
We are currently engaged exclusively in the clinical development of Apitox for continued use in knee OA through a Phase III trial in knee OA and potential use for MS through the early prosecution of appropriate patient populations through non-registered corporate sponsorship studies.
Sales and Marketing
The healthcare providers associated with the treatment of inflammation and pain management symptoms associated with OA and MS are not limited to one specialist but involve a comprehensive team of providers focused on slowing the progression of the disease along with the physical, emotional and day-to-day management of the condition. Each of these providers represents a potential customer for Apitox.
Apitoxin, which will be known as Apitox in the United States, has established technological credibility through its preclinical testing, Phase I, Phase II and preliminary Phase III clinical studies completed by Apimeds Korea. Apimeds Korea received regulatory approval for Apitoxin by the MFDA in South Korea, as well as long-term safety data from treatment of patients in Korea from 2003 to 2009. There were no serious adverse events from over 3,000 patients monitored, and Apitoxin has been approved and marketed in South Korea for OA since 2003. We update the FDA annually on safety data generated by Apimeds Korea from South Korea.
We aim to obtain FDA approval for Apitox in the United States market for treatment of inflammation and pain management symptoms associated with knee OA, and eventually MS, and expand the indication portfolio in the autoimmune market with a strategic marketing partner. The marketing partner strategy is common in the pharmaceutical marketplace, as the infrastructure, overhead, and barriers to entry dilute the focus and can rapidly erode the financial well-being of small, product development-based companies such as us. By identifying the strategic marketing partner at an early stage, the companies can deliver a final product, or family of products, in a form factor or variety of form factors over time, that specifically suit the target market. We believe that Apitox represents a significant opportunity as a platform technology, with numerous product-line extensions, and the potential for new, ancillary products such as delivery devices.
9
Reimbursement Strategy
Apimeds expects to apply to the Centers for Medicare and Medicaid Studies (“CMS”) for temporary generic reimbursement codes 12 to 18 months prior to a BLA approval. Temporary codes are used until manufacturers apply for, and receive, permanent codes, which identify the drug and its therapeutic class. Permanent codes are issued by CMS on a rolling quarterly basis.
We will engage third party contractors to assist the us with reimbursement, coding and policy development prior to, during and at the time of approval of Apitox. We will look for a contractor to provide the following services to us:
| ● | Coding Assessment and Strategy/Execution — CPT Review of Apitox Administration by Multiple Intradermal Injections. Assess the landscape to ensure a clear understanding of the key dynamics and analyze relevant proxies and precedent. Further assess relevant drug administration codes and whether appropriate codes exist. |
| ● | Medical Coverage Policy Analysis — Provide a framework and set expectations for Medicare’s anticipated coverage approach to Apitox, specifically in the context of intra articular hyaluronic acid use agent coverage policies and implications of their efficacy uncertainty. |
| ● | Medicare Local Coverage Analysis and Implications — Given the significance of Medicare policy standards, local and national Medicare policies often shape payer and provider perceptions and decisions. As complex statutory and regulatory guidance shape Medicare decision-making, ADVI analyzes, investigates, and synthesize Medicare policies that could affect access (coverage, coding and reimbursement) for Apitox. |
| ● | Medicaid and Commercial Coverage Analysis and Implications — Analyze available medical policies for five large state Medicaid agencies (based on population and geographic variation) and major commercial payers (where publicly available). |
| ● | Payer Policy Internal Expert Interviews — Conduct payer interviews with relevant Medicare, Medicaid and commercial policy advisors. |
| ● | HCPCS Coding and Payment Assessment — Assess the coding and reimbursement landscape to ensure Apimeds has a clear understanding of the key dynamics with the HCPCS application process and the Medicare Hospital Outpatient Prospective Payment System (OPPS) pass-through status application process. Through this assessment, identify the areas of concern, expectations, timing, timelines, and processes associated. This is especially relevant given the 2020 implementation of a new HCPCS review process. |
| ● | Address key Part B/medical benefit implications to Apitox in the following fields: |
| ● | HCPCS and OPPS application timelines (and potential evolution leading to launch). |
| ● | Coding/access implications prior to code assignment (e.g., NOC/miscellaneous codes), review the merits/risks of Q-code. |
| ● | further review the application processes, expectations, case examples, timelines, and hurdles that APUS may face across settings of care, payers, and with CMS, |
| ● | Case examples, timelines, and hurdles across settings of care with payers and CMS, |
| ● | Review of reimbursement implications; and |
| ● | Methodologies (ASP, WAC, AWP), role of sequestration, 340B, patient financial burden |
| ● | Develop Payer (with Emphasis on Medicare) Launch Recommendations — Based on the above primary and secondary research, synthesize the discussions and summarize the overall findings of the payer survey, highlighting themes, and provide recommendations and considerations for optimizing market access, given the current and evolving reimbursement landscape. This section will include payer (emphasis on Medicare) launch strategy recommendations (including timeline) and a local/national Medicare engagement strategy. |
10
Competition
We compete in an industry characterized by rapidly advancing technologies, intense competition, a changing regulatory and legislative landscape and a strong emphasis on the benefits of intellectual property protection and regulatory exclusivities.
Like any biopharmaceutical company, we face competition from multiple sources, including large or established pharmaceutical, biotechnology, and wellness companies, academic research institutions, government agencies, and private institutions. We believe our drug candidate will prevail amid the competitive landscape through its efficacy, safety, administration methods, cost, public and institutional demand, intellectual property portfolio, and treatment of the root cause of many age-associated diseases.
Many of our competitors, either alone or with strategic partners, have substantially greater financial, technical, and human resources than we do. Accordingly, our competitors may be more successful in obtaining approval for treatments and achieving widespread market acceptance, rendering our treatments obsolete or non-competitive. Accelerated merger and acquisition activity in the biotechnology and biopharmaceutical industries may result in even more resources concentrated among a smaller number of our competitors. These companies also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical study sites, patient registration for clinical studies, and acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunity could be substantially limited in the event that our competitors develop and commercialize products that are more effective, safer, more tolerable, more convenient, or less expensive than our comparable products. In geographies that are critical to our commercial success, competitors may also obtain regulatory approvals before us, resulting in our competitors building a strong market position in advance of our products’ entry. We believe the factors determining the success of our programs will be the efficacy, safety, and convenience of our drug candidates.
Additionally, consumer preference for branded, generic or private label products sold by competitors could adversely impact our financial performance. Our competitors, which differ within individual geographic markets, include large-scale retailers, smaller high-growth companies (which often operate on a regional basis and offer aggressive competition), multinational corporations moving into or expanding their presence in the consumer healthcare market, and “private-label” products sold by retailers.
Our aim is to reduce the use of NSAIDS and opioid use as it relates to the pain management associated with OA. We believe that if approved by the FDA, Apitox may be a non-addictive option to patients experiencing debilitating pain.
Business Agreement
On August 2, 2021, we entered into an agreement with Apimeds Korea, a principal stockholder of the Company (the “Business Agreement”). Pursuant to the Business Agreement, Apimeds Korea granted to the Company a sublicensable, royalty-bearing license to utilize all prior clinical development data associated with Apitoxin, Apitox, and all related names, advance clinical research, develop, manufacture and commercialize and sell Apitox in the United States. In exchange for this license, the Company will pay Apimeds Korea a perpetual royalty of 5% of the Company’s earnings before interest and taxes (as determined consistent with GAAP, derived from the sale or license of Apitox, less any shipping, handling, and insurance charges, credits (arising from returns or other adjustments), discounts, rebates, or allowances of any kind (if any). The Business Agreement can be terminated by mutual written agreement by the parties and will automatically terminate upon the bankruptcy or dissolution of the Company.
Assignment Agreement
On October 12, 2021, we entered into an intellectual property assignment agreement (the “Assignment Agreement”), which was effective as of May 12, 2020, with Apimeds Korea and Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and the founder of Apimeds Korea. During Dr. Kim’s engagement with Apimeds Korea, he contributed to the development of the intellectual property as it relates to Apitoxin, which will be marketed in the United States as Apitox (the “Assigned IP”).
11
Pursuant to the Assignment Agreement, Dr. Kim sold, transferred, and conveyed all his rights, title and interest in the Assigned IP to Apimeds Korea. Dr. Kim retained no right to use the Assigned IP. Additionally, the Assignment Agreement acknowledged that the Assigned IP was licensed to us to use via the Business Agreement.
Intellectual Property
Apitox’s API is bee venom, a natural, non-synthetic compound that is not patentable, so we rely principally on trade secrets to protect our rights to Apitox, particularly the method and process of manufacturing Apitox.
Supplier
We purchase venom from our United States supplier, Apico, Inc. (“Apico”), via a letter agreement. Pursuant to the letter agreement, Apico agreed that for a period of ten years, or until November 3, 2031 it would not supply Apis Mellifea venom for pharmaceutical use for any buyer other than us; provided that Apico may also supply Apimeds Korea for its use outside of the United States. The letter agreement excludes customers using venom for immunology, cosmetic or any other “non-pharmaceutical” use. The letter agreement may be terminated upon mutual written consent of both Apico and the Company.
Apico has developed and practices a proprietary method of harvesting venom. It operates under and is certified in current good manufacturing practice regulations enforced by the FDA and has an active and current Drug Master File (“DMF”) with the FDA. DMF’s are submissions to the FDA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of human drug products. We have an exclusive relationship with our supplier for pharmaceutical use in the United States and they are not permitted to sell to any other party for pharmaceutical use.
Apimeds Korea has a number of proprietary analytical methods for the classification and identification of specific pharmacologically active fractions of its venom, along with numerous manufacturing processes from filtration, vial filing and lyophilization required to produce Apitoxin. Apitoxin is the only approved and commercially available therapeutic product containing purified and sterile bee venom that is registered as an API in South Korea. The proprietary methods developed and practiced for the commercial manufacturing of Apitoxin include dilution, filtering, vial staging and lyophilization parameters and cycles.
We plan to file Apitox as a BLA with the Centers for Biologics and Research of the FDA following the successful completion of our Phase III trial for knee OA. The FDA provides 12-year market exclusivity at the time of approval of a BLA, with the potential for a six-month extension upon approval for pediatric use. If the BLA is approved, the 12-year period would be retroactive to the date of the application.
We intend to file a U.S. trademark application for “Apitox”.
Regulatory Environment
Government Regulation and Product Approval
In the United States, biological products are subject to regulation under the Federal Food, Drug, and Cosmetic Act (the “FDCA”), and the Public Health Service Act (the “PHSA”), and other federal, state, and local statutes and regulations. Both the FDCA and PHSA and their corresponding regulations govern, among other things, the research, development, clinical trials, testing, manufacturing, quality control, safety, purity and potency (efficacy), labeling, packaging, storage, record keeping, distribution, reporting, marketing, promotion, advertising, post-approval monitoring, and post-approval reporting involving biological products. Along with third-party contractors, we will be required to navigate the various preclinical and clinical regulatory obligations and the commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidate. The processes for obtaining regulatory approvals in the United States, along with subsequent compliance with applicable laws and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
12
Government policies may change, and additional government regulations may be enacted that could prevent or delay further development or regulatory approval of any product candidates, product or manufacturing changes, additional disease indications or label changes. We cannot predict the likelihood, nature or extent of government regulation that might arise from future legislative or administrative action.
Review and Approval for Licensing Biologics in the United States
In the United States, FDA regulates our current product candidate as a biological product, or biologics, under the FDCA, the PHSA, and associated implementing regulations. Biologics, like other drugs, are used for the diagnosis, cure, mitigation, treatment, or prevention of disease in humans. In contrast to low molecular weight drugs, which have a well-defined structure and can be thoroughly characterized, biologics are generally derived from living material (human, animal, or microorganism), are complex in structure, and thus are usually not fully characterized.
Biologics are also subject to other federal, state, and local statutes and regulations. The failure to comply with applicable statutory and regulatory requirements at any time during the product development process, approval process, or after approval may subject a sponsor or applicant to administrative or judicial enforcement actions. These actions could include the suspension or termination of clinical trials by FDA, FDA’s refusal to approve pending applications or supplemental applications, withdrawal of an approval, issuance of warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, import detention, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by FDA, the Department of Justice (“DOJ”), and other governmental entities.
An applicant seeking approval to market and distribute a biologic in the United States must typically undertake the following:
| ● | completion of non-clinical laboratory tests and studies performed in accordance with FDA’s good laboratory practice (“GLP”) regulations; |
| ● | manufacture, labeling and distribution of investigational drugs in compliance with FDA’s current good manufacturing practice (“cGMP”) requirements; |
| ● | submission to FDA of an investigational new drug application (“IND”), which must become effective before clinical trials may begin and must be updated annually and when significant changes are made; |
| ● | approval by an independent institutional review board (“IRB”) for each clinical site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with FDA’s Good Clinical Practices (“GCP”) to establish the safety, purity, and potency of the proposed biological product candidate for its intended purpose; |
| ● | after completion of all pivotal clinical trials, preparation of and submission to FDA of a BLA requesting marketing approval, which includes providing sufficient evidence to establish the efficacy, safety, purity, and potency of the proposed biological product for its intended use, including from results of nonclinical testing and clinical trials; |
| ● | satisfactory completion of an FDA advisory committee review, when appropriate, as may be requested by FDA to assist with its review; |
| ● | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities at which the proposed product, or certain components thereof, are produced to assess compliance with cGMP and data integrity requirements to assure that the facilities, methods, and controls are adequate to preserve the biological product’s identity, strength, quality, and purity and, if applicable, FDA’s good tissue practice (“GTP”) requirements for human cellular and tissue products; |
| ● | satisfactory completion of FDA inspections of selected clinical investigation sites to assure compliance with GCP requirements and the integrity of the clinical data; |
| ● | satisfactory completion of an FDA sponsor GCP inspection, often conducted at the applicant’s headquarters facility; |
13
| ● | payment of user fees (unless there is a waiver, exemption, or reduction) under the Prescription Drug User Fee Act (“PDUFA”) for the relevant year; |
| ● | FDA’s review and approval of the BLA to permit commercial marketing of the licensed biologic for particular indications for use in the United States; |
| ● | compliance with post-approval requirements, including the potential requirements to implement a risk evaluation and mitigation strategy (“REMS”), to report adverse events and biological product deviations, and to complete any post-approval studies; and |
| ● | completion of any post-approval clinical studies required by FDA, such as confirmatory trials or pediatric studies. |
From time to time, legislation is drafted, introduced, and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing, and marketing of biological products regulated by FDA. In addition to new legislation, FDA regulations, guidance documents, and policies are often revised or interpreted by the agency in ways that may significantly affect the regulation of biological products in the United States. It is impossible to predict whether further legislative changes will be enacted or whether FDA regulations, guidance, policies, or interpretations will change, and the effects of any such changes.
Preclinical and Clinical Development
Before an applicant can begin testing the potential product candidate in human subjects, the applicant must first conduct preclinical studies. Preclinical studies may include laboratory evaluations of product chemistry, toxicity, and formulation, as well as in vitro and animal studies to assess the potential safety and activity of the drug for initial testing in humans and to establish a rationale for therapeutic use. Preclinical studies are subject to federal regulations and requirements, including GLP regulations, which govern the conduct of animal studies designed to test a product’s safety. None of our preclinical studies to date have been animal studies. The results of an applicant’s preclinical studies are submitted to FDA as part of an IND.
An IND is a request for authorization from FDA to administer an investigational new drug product to humans. An IND is an exemption from the FDCA that allows an unapproved drug to be shipped in interstate commerce for use in a clinical trial. Such authorization must be secured prior to interstate shipment and administration of a biological drug that is not subject of an approved BLA. In support of an IND, applicants must submit a protocol for each clinical trial, which details, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments.
Human clinical trials may not begin until an IND is effective. The IND automatically becomes effective 30 days after receipt by FDA, unless FDA raises safety concerns or questions about the proposed clinical trial within the 30-day time period. In such a case, FDA may place the IND on clinical hold and the IND sponsor must resolve any of FDA’s outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in regulatory authorization to begin a clinical trial.
FDA may also place a clinical hold or partial clinical hold on a clinical trial following commencement of the trial under an IND. A clinical hold is an order issued by FDA to the sponsor to delay a proposed clinical investigation or to suspend an ongoing investigation. A partial clinical hold is a delay or suspension of only part of the clinical work requested under the IND. For example, under a partial clinical hold, FDA may instruct a sponsor not to enroll any new patients into a study but permit the previously enrolled patients to continue in the study. No more than 30 days after imposition of a clinical hold or partial clinical hold, FDA will provide the sponsor a written explanation of the basis for the hold. Following issuance of a clinical hold or partial clinical hold, an investigation may only resume after the FDA has notified the sponsor that the investigation may proceed. FDA will base that determination on information provided by the sponsor addressing the deficiencies previously cited or otherwise satisfying FDA that the investigation can proceed.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCP regulations, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. If a sponsor chooses to conduct a foreign clinical study under an IND, all FDA IND requirements must be met unless waived. When the foreign clinical study is not conducted under an IND, the sponsor must ensure that the study complies with GCP regulations in order to use the study as support for an IND or application for marketing approval, including review and approval by an IRB and informed consent from subjects.
14
Furthermore, an independent IRB for all sites participating in a clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at each site and must monitor the trial until completed. Regulatory authorities, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives.
Some trials also include oversight by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board (“DSMB”). DSMBs review unblinded study data at pre-specified times during the course of the study. If the DSMB determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy, the DSMB can make a recommendation to the sponsor to modify or stop the trial.
Other grounds for a sponsor’s decision to suspend or terminate a study may be made based on evolving business objectives or the competitive climate.
For purposes of BLA approval, clinical trials are typically conducted in the following sequential phases:
| ● | Phase 1: The investigational product is initially introduced into a small group of healthy human subjects or patients with the target disease or condition. These trials are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans and the side effects associated with increasing doses. These trials may also yield early evidence of effectiveness. |
| ● | Phase 2: The investigational product is administered to a slightly larger patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages, and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase III clinical trials. |
| ● | Phase 3: The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to generate sufficient data to statistically demonstrate the efficacy and safety of the product, to establish the overall risk/benefit ratio of the investigational product, and to provide an adequate basis for product approval by FDA. |
These phases may overlap or be combined. In some cases, FDA may require, or companies may voluntarily pursue, additional clinical trials after a product are approved to gain more information about the product, referred to as Phase 4 trials. Post-approval trials are conducted following initial approval, often to develop additional data and information relating to the use of the product in new indications.
Progress reports detailing the results of the clinical trials must be submitted at least annually to FDA. In addition, IND safety reports must be submitted to FDA for any of the following: serious and unexpected suspected adverse reactions in study subjects; findings from epidemiological studies, pooled analysis of multiple studies, animal or in vitro testing, or other clinical studies, whether or not conducted under an IND, and whether or not conducted by the sponsor, that suggest a significant risk in humans exposed to the drug; and any clinically important increase in the rate of a serious suspected adverse reaction over such rate listed in the protocol or investigator brochure.
A sponsor’s planned clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. FDA will typically inspect one or more clinical sites to assure compliance with GCP and the integrity of the clinical data submitted.
15
During clinical development, the sponsor often refines the indication and endpoints on which the BLA will be based. For endpoints based on patient-reported outcomes (“PROs”), the process typically is an iterative one. FDA has issued guidance on the framework it uses to evaluate PRO instruments. Although the agency may offer advice on optimizing PRO instruments during the clinical development process, FDA usually reserves final judgment until it reviews the BLA.
Concurrent with clinical trials, companies often complete additional animal studies, and develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, must develop methods for testing the identity, strength, quality, purity and potency of the final drug. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
BLA Submission and Review
Assuming successful completion of all required clinical testing in accordance with all applicable regulatory requirements, an applicant may submit a BLA requesting licensing to market the biologic for one or more indications in the United States. The BLA must include the results of nonclinical studies and clinical trials; detailed information on the product’s chemistry, manufacture, controls; and proposed labeling. Under the PDUFA, a BLA submission is subject to an application user fee, unless a waiver, reduction, or exemption applies.
FDA will initially review the BLA for completeness before accepting it for filing. Under FDA’s procedures, the agency has 60 days from its receipt of a BLA to determine whether the application will be accepted for filing and substantive review. If the agency determines that the application does not meet this initial threshold standard, FDA may refuse to file the application and request additional information, in which case the application must be resubmitted with the requested information and review of the application delayed.
After the BLA is accepted for filing, FDA reviews the BLA to determine, among other things, whether a product is safe, pure, and potent and if the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued identity, strength, quality, safety, purity, and potency. To ensure cGMP, GLP, GCP, GTP, and other regulatory compliance, an applicant must incur significant expenditure of time, money, and effort in the areas of training, record keeping, production and quality control. In addition, FDA expects that all data be reliable and accurate, and requires sponsors to implement meaningful and effective strategies to manage data integrity risks. Data integrity is an important component of the sponsor’s responsibility to ensure the safety, efficacy and quality of its product or products.
For cellular products, FDA will not approve the product if the manufacturer is not in compliance with the GTPs, to the extent applicable. GTPs are FDA regulations and guidance documents that govern the methods used in, and the facilities and controls used for, the manufacture of human cells, tissue, and cellular and tissue-based products (“HCT/Ps”), which are human cells or tissue intended for implantation, transplant, infusion, or transfer into a human recipient. The primary intent of the GTP requirements is to ensure that cell and tissue-based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. FDA regulations also specify how HCT/P establishments must register and list their HCT/Ps with FDA and how they must evaluate donors through screening and testing, where applicable.
If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The performance goals and policies implemented by FDA under the PDUFA generally provide for FDA action on an original BLA within 10 months of filing, which (as discussed above) typically occurs within 60 days of submission, but that deadline is extended in certain circumstances. Furthermore, the review process is often significantly extended by FDA’s requests for additional information or clarification.
16
FDA may refer applications for novel products or products that present difficult questions of safety or efficacy to an advisory committee. Typically, an advisory committee consists of a panel that includes clinicians and other experts who will review, evaluate, and provide a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions and usually has followed such recommendations.
After FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its components will be produced, FDA may issue an approval letter or a Complete Response Letter (“CRL”). An approval letter authorizes commercial marketing of the biological with specific prescribing information for specific indications. A CRL will describe all of the deficiencies that FDA has identified in the BLA, except that where FDA determines that the data supporting the application are inadequate to support approval, FDA may issue the CRL without first conducting required inspections, testing submitted product lots and/or reviewing proposed labeling. If and when the deficiencies have been addressed to FDA’s satisfaction in a resubmission of the BLA, FDA will issue an approval letter. In issuing the CRL, the FDA may recommend actions that the applicant might take to place the BLA in condition for approval, including requests for additional data, information, or clarification. FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied and may require additional testing or information and/or require new clinical trials. Even with submission of this additional information, FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
During the approval process, FDA will determine whether a REMS is necessary to help ensure the benefits outweigh the risks of the biologic. A REMS is a safety strategy to manage a known or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. If FDA concludes that a REMS is needed, the BLA sponsor must submit a proposed REMS and FDA will not approve the BLA without a REMS that the agency has determined is acceptable.
If the FDA approves a product, it may limit the approved indications for use for the product, or require that contraindications, warnings, or precautions be included in the product labeling. FDA may also require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess the drug’s safety after approval. FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs.
FDA may also require testing and surveillance programs to monitor the product after commercialization. For biologics, such testing may include official lot release, which requires the manufacturer to perform certain tests on each lot of the product before it is released for distribution. The manufacturer then typically must submit samples of each lot of products to the FDA, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot. The FDA may also perform certain confirmatory tests on lots of some products itself, before releasing the lots for distribution by the manufacturer.
In general, an approved BLA only allows the sponsor to market the biologic as approved, without modification. If, for example, a sponsor modifies an approved T cell product to target different peptides or in our case to target another HLA type, the sponsor would be required to either file a supplemental BLA with FDA or receive FDA approval for a comparability protocol in order to implement this change into the final product.
The FDA may withdraw the product approval if compliance with pre- and post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace.
Post-Approval Requirements
Any products manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, reporting of certain deviations and adverse experiences, product sampling and distribution, and advertising and promotion of the product. After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are often subject to further testing requirements and FDA review and approval, depending on the nature of the post-approval change. There also are continuing user fee requirements, under which FDA assesses an annual program fee for each product identified in an approved BLA. Biologic manufacturers and their third-party contractors are required to register their facilities with the FDA and certain state agencies. These facilities are subject to routine and periodic unannounced inspections by FDA and certain state agencies for compliance with cGMP, post-marketing safety reporting and data integrity requirements, which impose certain procedural and documentation requirements to assure quality of manufacturing and product. FDA has increasingly observed cGMP violations involving data integrity during site inspections and is a significant focus of its oversight. Requirements with respect to data integrity include, among other things, controls ensuring complete and secure data; activities documented at the time of performance; audit trail functionality; authorized access and limitations; validated computer systems; and review of records for accuracy, completeness, and compliance with established standards.
17
Post-approval changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon the sponsor and any third-party manufacturers that the sponsor may use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain compliance with cGMP, data integrity, pharmacovigilance, and other aspects of regulatory compliance.
The FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-approval studies to assess new safety risks; or imposition of distribution or other restrictions under a REMS. Other potential consequences include, for example:
| ● | restrictions on the marketing or manufacturing of a product, complete withdrawal of the product from the market, or product recalls; |
| ● | fines, warning or untitled letters, or holds on post-approval clinical studies; |
| ● | refusal of FDA to approve pending applications or supplements to approved applications, or suspension or revocation of existing product approvals; |
| ● | product seizure or detention, or refusal of FDA to permit the import or export of products; or |
| ● | permanent injunctions and consent decrees, including the imposition of civil or criminal penalties. |
FDA strictly regulates the marketing, labeling, advertising, and promotion of prescription drug products placed on the market. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved labeling. FDA’s regulation includes, among other things, standards and regulations for direct-to-consumer advertising, communications regarding unapproved uses, industry-sponsored scientific and educational activities and promotional activities involving the Internet and social media. Promotional claims relating to a product’s safety or effectiveness are prohibited before the drug is approved. After approval, a product generally may not be promoted for uses that are not approved by FDA, as reflected in the product’s prescribing information. In the United States, healthcare professionals are generally permitted to prescribe drugs for such uses not described in the drug’s labeling, known as off-label uses, because FDA does not regulate the practice of medicine. However, FDA regulations impose rigorous restrictions on manufacturers’ communications and prohibit the promotion of off-label uses. It may be permissible, under very specific, narrow conditions, for a manufacturer to engage in non-promotional, non-misleading communication regarding off-label information, such as distributing scientific or medical journal information.
If a company is found to have promoted off-label uses, it may become subject to adverse public relations and administrative and judicial enforcement by FDA, the DOJ, or the Office of the Inspector General of the Department of Health and Human Services (“HHS”), as well as other federal and state authorities. This could subject a company to a range of penalties that could have a significant commercial impact, including civil, administrative, and criminal fines, penalties, and agreements that materially restrict the manner in which a company promotes or distributes products. The federal government has levied large civil, administrative, and criminal fines and penalties against companies for alleged improper promotion and has also requested that companies enter into Corporate Integrity Agreements and Consent Decrees of Permanent Injunction under which specified promotional conduct is changed or curtailed.
18
The distribution of prescription drugs and biologics are subject to the Drug Supply Chain Security Act (“DSCSA”), which requires manufacturers and other stakeholders to comply with product identification, tracing, verification, detection and response, notification, and licensing requirements. In addition, the Prescription Drug Marketing Act and its implementing regulations and state laws limit the distribution of prescription pharmaceutical product samples, and the DSCSA imposes requirements to ensure accountability in distribution and to identify and remove prescription drug and biological products that may be counterfeit, stolen, contaminated, or otherwise harmful from the market.
Expedited Development and Review Programs
FDA offers a number of expedited development and review programs for qualifying product candidates. The fast-track program is intended to expedite or facilitate the process of reviewing new products that meet certain criteria. Specifically, new products are eligible for fast-track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. A product intended to treat a serious or life-threatening disease or condition may also be eligible for breakthrough therapy designation to expedite its development and review. Any marketing application for a biologic submitted to FDA for approval, including a product with a fast-track designation and/or breakthrough therapy designation, may be eligible for other types of FDA programs intended to expedite FDA review and approval process, such as priority review and accelerated approval. FDA also may grant accelerated approval to certain products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions.
The RMAT designation, which we are currently planning to seek for some of our therapies, is intended to facilitate an efficient development program for, and expedite review of, any drug that meets the following criteria: (1) the drug is a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with limited exceptions; (2) the drug is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (3) preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such a disease or condition. Like breakthrough therapy designation, RMAT designation provides potential benefits that include more frequent meetings with FDA to discuss the development plan for the product candidate and eligibility for rolling review and priority review. Products granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites (including through expansion to additional sites) so as to remove any likelihood of site-specific or investigator-specific bias on the evidence of effectiveness. Once approved, when appropriate, FDA can permit fulfillment of post-approval requirements for RMATs receiving accelerated approval through the submission of clinical evidence, clinical studies, patient registries, or other sources of real-world evidence such as electronic health records; through the collection of larger confirmatory datasets; or through post-approval monitoring of all patients treated with the therapy prior to approval.
Fast track designation, breakthrough therapy designation, priority review, accelerated approval, and RMAT designation do not change the standards for approval but may expedite the development or approval process.
Patent Term Restoration and Marketing Exclusivity
After approval, owners of relevant drug or biological product patents may apply for up to a five year term patent extension to restore a portion of patent term lost during product development and FDA review of a BLA if approval of the application is the first permitted commercial marketing or use of a drug or biologic containing the active ingredient under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The allowable patent term extension is calculated as one-half of the product’s testing phase, which is the time between the effective date of an IND and initial BLA submission, and all of the approval phase, which is the time between BLA submission and approval, up to a maximum of five years. The time can be shortened if the FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years from the date of FDA approval of the product. Only one patent claiming each approved product is eligible for restoration and the patent holder must apply for restoration within 60 days of approval, even if the product cannot be commercially marketed at that time. The USPTO, in consultation with FDA, reviews and approves the application for patent term restoration.
For patents that might expire during the BLA application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the USPTO must determine that approval of the product candidate covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a product candidate for which a BLA has not been submitted.
19
Biosimilars and Marketing Exclusivities
The Biologics Price Competition and Innovation Act (“BPCIA”) created an abbreviated approval pathway for biological product candidates shown to be highly similar to or interchangeable with an FDA licensed biological product. A biological product on which another biological product candidate’s BLA relies to establish bio similarity is known as a reference product. Bio similarity sufficient to reference a prior FDA-approved product requires that there be no differences in conditions of use, route of administration, dosage form and strength, and no clinically meaningful differences between the biological product candidate and the reference product in terms of safety, purity, and potency. Bio similarity must be shown through analytical trials, animal trials and at least one clinical trial, unless the Secretary of HHS waives a required element. A biosimilar product candidate may be deemed interchangeable with a prior approved product if it meets the higher hurdle of demonstrating that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biological product candidate and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. Complexities associated with the larger, and often more complex, structures of biologics, as well as the process by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being resolved by FDA.
A reference biologic is granted 12 years of exclusivity from the time of first licensure of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biological product candidate submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against a finding of interchangeability for other biologics for the same condition of use for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) 18 months after the first interchangeable biosimilar is approved if there is no patent challenge, (iii) 18 months after resolution of a lawsuit over the patents of the reference biologic in favor of the first interchangeable biosimilar applicant, or (iv) 42 months after the first interchangeable biosimilar’s application has been approved if a patent lawsuit is ongoing within the 42 month period. At this time, it is unclear whether products deemed “interchangeable” by FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy laws and regulations.
Healthcare Regulation
Coverage, Pricing, and Reimbursement
Our ability to successfully commercialize any products for which we receive regulatory approval for commercial sale will depend, in part, on the extent to which third-party payors provide coverage and establish adequate reimbursement levels for such products, and significant uncertainty exists as to the coverage and reimbursement status of any products for which may we obtain regulatory approval. In the United States, third-party payors include federal and state health care programs, private managed care providers, health insurers and other organizations. The process for determining whether a third-party payor will provide coverage for a product may be separate from the process for setting the price of a product or for establishing the reimbursement rate that such a payor will pay for the product. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication. Third-party payors are increasingly challenging the price, examining the medical necessity, and reviewing the cost-effectiveness of medical products, therapies, and services, in addition to questioning their safety and efficacy. We may need to conduct expensive pharmaco-economic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our product candidates may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
20
The marketability of any product candidates for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on healthcare pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Compliance Requirements
Although we currently do not have any commercialized products, our current and future business operations may be subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security, price reporting and physician sunshine laws. Some of our pre-commercial activities are subject to some of these laws.
The federal Anti-Kickback Statute makes it illegal for any person or entity, including a prescription drug manufacturer or a party acting on its behalf to knowingly and willfully, directly or indirectly, solicit, receive, offer, or pay any remuneration in cash or in kind that is intended to induce or reward the referral of business, including the purchase, order, or lease of any item or service for which payment may be made under a federal healthcare program, such as Medicare or Medicaid. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, formulary managers and beneficiaries on the other.
Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have found that the Anti-Kickback Statute may be violated if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare program business. In addition, liability may be established without actual knowledge of the statute or specific intent to violate it. Violations of this law are punishable by up to ten years in prison, and can also result in criminal fines, civil money penalties and exclusion from participation in federal healthcare programs.
Moreover, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
The federal civil False Claims Act prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment of government funds or knowingly making, using, or causing to be made or used, a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. Persons and entities can be held liable under these laws if they are deemed to “cause” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. Many pharmaceutical and other healthcare companies have been investigated and have reached substantial financial settlements with the federal government under the civil False Claims Act for a variety of alleged improper marketing activities, including: providing free product to customers with the expectation that the customers would bill federal programs for the product; providing sham consulting fees, grants, free travel and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which are used to set drug payment rates under government healthcare programs. Penalties for federal civil False Claims Act violations may include up to three times the actual damages sustained by the government, plus mandatory civil penalties of between $13,508 and $27,018 for each separate false claim, and the potential for exclusion from participation in federal healthcare programs. In addition, although the federal False Claims Act is a civil statute, False Claims Act violations may also implicate various federal criminal statutes.
The healthcare fraud provisions of the Health Insurance Portability and Accountability Act (“HIPAA”) prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
21
Many states have analogous laws and regulations, such as: state anti-kickback and false claims laws that may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to certain healthcare providers; laws that require drug manufacturers to report information related to clinical trials or information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; laws that restrict the ability of manufacturers to offer co-pay support to patients for certain prescription drugs; and laws and local ordinances that require identification or licensing of sales representatives.
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their implementing regulations, mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common healthcare transactions, as well as standards relating to the privacy and security of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. Among other things, HITECH makes HIPAA’s security standards directly applicable to business associates, defined as independent contractors or agents of covered entities that create, receive, or obtain protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities and business associates and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties.
The U.S. federal Physician Payment Sunshine Act, implemented as the Open Payments Program, requires manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to CMS information related to direct or indirect payments and other transfers of value to physicians and teaching hospitals (and certain other practitioners as of 2022), as well as ownership and investment interests held in the company by physicians and their immediate family members.
Because we intend to commercialize products that could be reimbursed under a federal health care program and other governmental healthcare programs, we intend to develop a comprehensive compliance program that establishes internal control to facilitate adherence to the rules and program requirements to which we will or may become subject. Although the development and implementation of compliance programs designed to establish internal control and facilitate compliance can mitigate the risk of investigation, prosecution, and penalties assessed for violations of these laws, the risks cannot be entirely eliminated.
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and individual imprisonment, any of which could adversely affect our ability to operate our business and our financial results
Health Care Reform
In the United States and some foreign jurisdictions, there have been, and continue to be, legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities, and affect the ability to profitably sell product candidates for which marketing approval is obtained. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
22
For example, the Affordable Care Act (“ACA”) substantially changed the way healthcare is financed by both the government and private insurers, and significantly impacts the U.S. pharmaceutical industry. The ACA contains provisions that may reduce the profitability of drug products through increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. The ACA made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate. The ACA also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and by enlarging the population potentially eligible for Medicaid drug benefits.
There have been judicial challenges to certain aspects of the ACA, as well as efforts by Congress to modify, and by agencies to alter the implementation of, certain aspects of the ACA. For example, Congress eliminated the tax penalty for failure to comply with the ACA’s individual mandate to carry health insurance. Further, the Bipartisan Budget Act of 2018, among other things, amended the ACA to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D to close the coverage gap in most Medicare drug plans, commonly referred to as the donut hole.
It is possible that the ACA, as currently enacted or as may be amended in the future, as well as other healthcare reform measures, including those that may be adopted in the future, may result in more rigorous coverage criteria, and less favorable payment methodologies, or other downward pressure on coverage and payment and the price that we receive for any approved product. Any reduction in reimbursement or restriction on coverage under Medicare or other federal health care programs may result in a similar reduction or restriction by private payors.
Other legislative changes have been proposed and adopted in the U.S. since the ACA was enacted. For example, the Inflation Reduction Act introduces several changes to the Medicare Part D benefit, including a limit on annual out-of-pocket costs and a change in manufacturer liability under the program which could negatively affect the profitability of our product candidates. The IRA sunsets the current Part D coverage gap discount program starting in 2025 and replaces it with a new manufacturer discount program. Failure to pay a discount under this new program will be subject to a civil monetary penalty. In addition, the IRA establishes a Medicare Part B inflation rebate scheme effective January 2023 and a Medicare Part D inflation rebate scheme effective October 2022, under which, generally speaking, manufacturers will owe rebates if the price of a Part B or Part D drug increases faster than the pace of inflation. Failure to timely pay a Part B or D inflation rebate is subject to a civil monetary penalty. The IRA also creates a drug price negotiation program under which the prices for Medicare units of certain high Medicare spend drugs and biologicals without generic or biosimilar competition will be capped by reference to, among other things, a specified non-federal average manufacturer price starting in 2026. Failure to comply with requirements under the drug price negotiation program is subject to an excise tax and/or a civil monetary penalty. Congress continues to examine various policy proposals that may result in pressure on the prices of prescription drugs with respect to the government health benefit programs and otherwise. The IRA or other legislative changes could impact the market conditions for our product candidates.
In general, there has been heightened governmental scrutiny over the manner in which drug manufacturers set prices for their commercial products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
23
Drug Pedigree Laws
State and federal governments have proposed or enacted various drug pedigree laws which can require the tracking of all transactions involving prescription drugs from the manufacturer to the pharmacy (or other dispensing) level. Companies are required to maintain records documenting the chain of custody of prescription drug products beginning with the purchase of such products from the manufacturer. Compliance with these pedigree laws requires implementation of extensive tracking systems as well as heightened documentation and coordination with customers and manufacturers. While we fully intend to comply with these laws, there is uncertainty about future changes in legislation and government enforcement of these laws. Failure to comply could result in fines or penalties, as well as loss of business that could have a material adverse effect on our financial results.
Federal Regulation of Patent Litigation Settlements and Authorized Generic Arrangements
As part of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, companies are required to file with the U.S. Federal Trade Commission (“FTC”) and the U.S. Department of Justice certain types of agreements entered into between brand and generic pharmaceutical companies related to the settlement of patent litigation or manufacture, marketing and sale of generic versions of branded drugs. This requirement could affect the manner in which generic drug manufacturers resolve intellectual property litigation and other disputes with brand pharmaceutical companies and could result generally in an increase in private-party litigation against pharmaceutical companies or additional investigations or proceedings by the FTC or other governmental authorities.
Other
The U.S. federal government, various states and localities have laws regulating the manufacture and distribution of pharmaceuticals, as well as regulations dealing with the substitution of generic drugs for branded drugs. Our operations are also subject to regulation, licensing requirements and inspection by the states and localities in which our operations are located or in which we conduct business.
Certain of our activities are also subject to FTC enforcement actions. The FTC enforces a variety of antitrust and consumer protection laws designed to ensure that the nation’s markets function competitively, are vigorous, efficient and free of undue restrictions. Federal, state, local and foreign laws of general applicability, such as laws regulating working conditions, also govern us.
In addition, we are subject to numerous and increasingly stringent federal, state and local environmental laws and regulations concerning, among other things, the generation, handling, storage, transportation, treatment and disposal of toxic and hazardous substances, the discharge of pollutants into the air and water and the cleanup of contamination. We are required to maintain and comply with environmental permits and controls for some of our operations, and these permits are subject to modification, renewal and revocation by the issuing authorities. Our environmental capital expenditures and costs for environmental compliance may increase in the future as a result of changes in environmental laws and regulations or increased manufacturing activities at any of our facilities. We could incur significant costs or liabilities as a result of any failure to comply with environmental laws, including fines, penalties, third-party claims and the costs of undertaking a clean-up at a current or former site or at a site to which our wastes were transported. In addition, we have grown in part by acquisition, and our diligence may not have identified environmental impacts from historical operations at sites we have acquired in the past or may acquire in the future.
Employees
As of the date of this Annual Report, we have two full time employees. We have no part-time employees and we engage one consultant. We believe that we maintain good relations with our employees.
Item 1A. Risk Factors
As a smaller reporting company, as defined in Rule 12b-2 of the Exchange Act, we are not required to provide the information required by this Item.
Item 1B. Unresolved Staff Comments
Not applicable.
24
Item 1C. Cybersecurity
Risk Management and Strategy
Managing Material Risks & Integrated Overall Risk Management
In the normal course of business, we may collect and store certain sensitive company information, including proprietary and confidential business information. We maintain various protections designed to safeguard against cyberattacks, including firewalls, key-based authentication, and virtual private networks. We protect against business interruption by backing up our major systems. We consider these cybersecurity risk management efforts as part of our broader risk management framework. This integration helps ensure that cybersecurity considerations are a fundamental part of our decision-making processes. Our management team works with our audit committee and board of directors (the “Board”) to evaluate and address cybersecurity risks in alignment with our business objectives and operational needs.
Engage Third Parties on Risk Management
To date, we have
Oversee Third Party Risk
We utilize various third-party software applications
in the functioning of our core business. We consider the cybersecurity practices of our
Risks from Cybersecurity Threats
We face risk from cybersecurity threats that could have a material adverse effect on our business, financial condition, results of operations, cash flows or reputation.
To date, we have not experienced any previous
cybersecurity incidents that have materially affected or are reasonably likely to
25
Governance
Board of Directors Oversight
Our Board is aware of the critical nature of managing risks associated with cybersecurity threats, and recognizes the significance of these threats to our operational integrity and stockholder confidence.
Risk Management Personnel
The audit committee is central to the Board’s
Management’s Role Managing Risk and Reporting to the Board
We do not currently have an employee who has significant and demonstrated professional IT management experience and possesses the requisite education, skills and experience needed to develop and execute our cybersecurity strategies. Presently, our senior management is responsible for monitoring our cybersecurity risks and maintaining an ongoing dialogue with the audit committee regarding emerging or potential cybersecurity risks as needed. The relationship between senior management and the audit committee regarding current and emerging cybersecurity concerns helps to integrate cybersecurity consideration into the Company’s broader strategic objectives.
Item 2. Properties
We are located at 2 East Broad Street, 2nd Floor, Hopewell, NJ 08525. This space is donated to us by one of our officers and we do not pay a monthly fee. We believe our current facilities are suitable for our current operations.
Item 3. Legal Proceedings
We are not currently subject to any legal proceedings. However, we may from time to time become a party to various legal proceedings arising in the ordinary course of our business.
Item 4. Mine Safety Disclosures
Not applicable.
26
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities Market Information
There is no established public trading market for our common stock currently, nor can we give any assurance that one will develop. The Company intends to seek to list its shares of common stock on a national securities exchange (an “Exchange Listing”). Any Exchange Listing is subject to future market conditions and there can be no assurance that any Exchange Listing will occur. Until any Exchange Listing, our common stock is not registered under the Securities Act of 1933, as amended (the “Securities Act”), or any state securities law, and will be restricted as to transfer by law.
Holders
As of the date of this Annual Report, there were 9 holders of record of our common stock.
Dividends
We have not paid any cash dividends on our common stock to date. It is the present intention of our Board to retain all earnings, if any, for use in our business operations and, accordingly, our Board does not anticipate declaring any dividends in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report.
Unregistered Sales of Equity Securities
None.
Item 6. [Reserved]
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our financial statements and related notes and other financial information included elsewhere in this Annual Report. This discussion and analysis and other parts of this prospectus contain forward-looking statements based upon our current plans and expectations that involve risks, uncertainties and assumptions, such as statements regarding our plans, objectives, expectations, intentions and beliefs. Our actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under the section entitled “Risk Factors” and elsewhere in this Annual Report. Please also see the section entitled “Special Note Regarding Forward-Looking Statements.”
Overview
Apimeds Pharmaceuticals US, Inc. is a clinical stage biopharmaceutical company that is in the process of developing Apitox, a proprietary intradermally administered bee venom-based toxin. Our primary focus is to advance Apitox in the treatment of inflammatory conditions in the United States, specifically osteoarthritis (“OA”) and, eventually, multiple sclerosis (“MS”).
Apitox, is currently marketed and sold by Apimeds, Inc. in South Korea (“Apimeds Korea”) as “Apitoxin” for the treatment of inflammation and pain management symptoms associated with OA. There is an extensive history of use of bee venom, both in the United States and around the world, to assist with pain management. We believe that, in addition to knee OA and MS, Apitox has the potential to help manage difficult to control pain and inflammation issues, which we will explore in the future.
27
Our Product Candidate
Our product candidate Apitox is a purified, pharmaceutical grade venom of the Apis mellifera, or honeybee, which is classified by the U.S Food and Drug Administration (“FDA”) as an active pharmaceutical ingredient (“API”). Apimeds Korea has developed a proprietary method and process of turning extracted bee venom into a lyophilized powder for reconstitution prior to intradermal dose injections, which they sell in Korea as South Apitoxin. Apimeds Korea has exclusively licensed to us all rights to develop, commercialize, market and sell Apitoxin as “Apitox” in the United States in exchange for a sales royalty. See “Item 13. Certain Relationships and Related Transactions, and Director Independence — Certain Relationships and Related Transactions — Business Agreement.”
The success of the Company is dependent on obtaining the necessary regulatory approvals of its product candidates, marketing its products and achieving profitable operations. The continuation of the research and development activities and the commercialization of its products, if approved, are dependent on the Company’s ability to successfully complete these activities and to obtain additional financing through a combination of financing activities and operations. It is not possible to predict either the outcome of future research and development or commercialization programs, or the Company’s ability to fund these programs.
Financial Results
Since inception, Apimeds has incurred significant operating losses. For the years ended December 31, 2024 and 2023, Apimeds Pharmaceuticals US, Inc. net loss was $1,389,990 and $777,694, respectively. As of December 31, 2024, Apimeds Pharmaceuticals US, Inc. had an accumulated deficit of $4,391,924, a stockholders’ deficit of $1,358,121 and a working capital deficit of $1,011,277.
Going Concern
The Company has evaluated whether there are any conditions and events, considered in the aggregate, that raise substantial doubt about its ability to continue as a going concern within one year beyond the issuance date of these financial statements. As of December 31, 2024, the Company had accumulated deficit amount to $4,391,924. The Company incurred net losses of $1,389,990 for the year ended December 31, 2024, and expects to continue to incur substantial losses in the future. Based on such conditions and the Company’s current plans, which are subject to change, management believes that the Company’s existing cash as of December 31, 2024, is not sufficient to satisfy its operating cash needs for 12 months from the issuance date of the report.
The accompanying financial statements have been prepared assuming the Company will continue to operate as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business, and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from uncertainty related to its ability to continue as a going concern.
The success of the Company is dependent on obtaining the necessary regulatory approvals of its product candidates, marketing its products and achieving profitable operations. The continuation of the research and development activities and the commercialization of its products, if approved, are dependent on the Company’s ability to successfully complete these activities and to obtain additional financing through a combination of financing activities and operations. If the Company is unable to maintain sufficient financial resources, its business, financial condition and results of operations will be materially and adversely affected. This could affect future development and business activities and potential future clinical studies and/or other future ventures. There can be no assurance that the Company will be able to obtain the needed financing on acceptable terms or at all.
28
Results of operations for the years ended December 31, 2024 and 2023
Operating Expense
The following table sets forth the Company’s selected statements of operations data for the following periods:
| Years Ended December 31, | ||||||||||||
| 2024 | 2023 | Change | ||||||||||
| Operating expenses | ||||||||||||
| Research and development expenses | $ | - | $ | 98,544 | $ | (98,544 | ) | |||||
| General and administrative expenses | 1,275,095 | 648,892 | 626,203 | |||||||||
| Loss from operations | (1,275,095 | ) | (747,436 | ) | (527,659 | ) | ||||||
| Other expenses | ||||||||||||
| Interest income | 2,824 | 7,811 | (4,987 | ) | ||||||||
| Interest expense | (117,719 | ) | (38,069 | ) | (79,650 | ) | ||||||
| Net loss | $ | (1,389,990 | ) | $ | (777,694 | ) | $ | (612,296 | ) | |||
Revenues
For the years ended December 31, 2024 and 2023, the Company had no revenue.
Research and Development Expenses
The following table summarizes the year-over-year changes in research and development expenses for the periods presented:
| Years Ended December 31, | ||||||||||||
| 2024 | 2023 | Change | ||||||||||
Research and development expenses |
$ | - | $ | 98,544 | $ | (98,544 | ) | |||||
| Total research and development expenses | $ | - | $ | 98,544 | $ | (98,544 | ) | |||||
Research and development expenses were $0 for the year ended December 31, 2024, compared to $98,544 for the same period in 2023, representing a decrease of $98,544. The decrease in research and development expenses was primarily attributed to a decrease as the Company was not performing any R&D activities currently in 2024.
General and administrative expenses
The following table summarizes the year-over-year changes in general and administrative expenses for the years presented:
| Years Ended December 31, | ||||||||||||
| 2024 | 2023 | Change | ||||||||||
| Payroll expenses | $ | 413,404 | $ | 114,000 | $ | 299,404 | ||||||
| Professional services | 815,271 | 485,949 | 329,322 | |||||||||
| Office expenses | 16,257 | 33,539 | (17,282 | ) | ||||||||
| General administrative | 30,163 | 15,404 | 14,759 | |||||||||
| $ | 1,275,095 | $ | 648,892 | $ | 626,203 | |||||||
General and administrative expenses were $1,2750,95 for the year ended December 31, 2024, compared to $648,892 for the same period in 2023, representing an increase of $626,203. The increase was mostly attributable to an increase in professional expenses for a total of approximately $329,000 and an increase in payroll expenses for the officers of the Company for a total of approximately $299,000.
29
Other Expense
The following table summarizes the year-over-year changes in general and administrative expenses for the years presented:
| Years Ended December 31, | ||||||||||||
| 2024 | 2023 | Change | ||||||||||
| Interest income | $ | 2,824 | $ | 7,811 | $ | (4,987 | ) | |||||
| Interest expense | (117,719 | ) | (38,069 | ) | (79,650 | ) | ||||||
| $ | (114,895 | ) | $ | (30,258 | ) | $ | (84,637 | ) | ||||
Other expense was $114,895 for the year ended December 31, 2024, compared to $30,258 for the same period in 2023. Representing an increase of $84,637. The increase was mainly due to an increase in interest expense for a total of approximately $80,000.
Net Loss
Net loss was $1,389,990 for the year ended December 31, 2024, compared to $777,694 in the same period of 2023, representing an increase of $612,296. The increase was mainly due to the increase in general and administrative expenses, specifically professional fees associated with the filing of the registration statement on Form S-1 with the U.S. Securities and Exchange Commission (the “SEC”) and pre-IPO expenses as well as an increase in payroll expenses.
Liquidity and Capital Resources
The Company has generated no revenue, has incurred operating losses since inception, expects to continue to incur significant operating losses for the foreseeable future and may never become profitable. Until such time as the Company is able to establish a revenue stream, it is dependent upon obtaining necessary equity and/or debt financing to continue operations. The Company cannot make any assurances that sales will commence in the near term or that additional financing will be available to it on acceptable terms or at all. This could negatively impact our business and operations and could also lead to the reduction of our operations.
Cash Flows
The following table presents selected financial information and statistics for each of the periods shown below:
| 2024 | 2023 | Change | ||||||||||
| Net cash used in operating activities | $ | (733,526 | ) | $ | (627,790 | ) | $ | (105,736 | ) | |||
| Net cash used in investing activities | - | - | - | |||||||||
| Net cash provided by financing activities | 326,500 | 1,032,100 | (705,600 | ) | ||||||||
| Net (decrease) increase in cash | $ | (407,026 | ) | $ | 404,310 | $ | (811,336 | ) | ||||
During the year ended December 31, 2024, operating activities used approximately $734,000 of cash, primarily resulting from a net loss of $1,389,990, partially offset by non-cash interest expense-related parties of $37,766, accretion expense of $79,953, and changes in operating assets and liabilities of $538,745.
During the year ended December 31, 2023, operating activities used approximately $628,000 of cash, primarily resulting from a net loss of $777,694, partially offset by stock compensation expense of $69,993, non-cash interest expense-related parties of $33,000, accretion expense of $5,069, and changes in operating assets and liabilities of $41,842.
30
Investing activities
During the years ended December 31, 2024 and 2023 investing activities used $0.
Financing activities
During the year ended December 31, 2024, financing activities provided $326,500 of cash resulting from $250,000 in proceeds from notes payable from related parties and cash advances from related parties of $76,500.
During the year ended December 31, 2023, financing activities provided $1,032,100 of cash resulting from $1,055,000 in proceeds from issuance of shares, cash advances from related parties of $9,000, offset by repayments to cash advances from related parties of $31,900.
Contractual Obligations and Commitments
See Note 4 – Debt, and Note 6 – Commitments and Contingencies, of the notes to the Company’s financial statements as of and for the year ended December 31, 2024 included elsewhere in this Annual Report for further discussion of the Company’s commitments and contingencies.
Off-Balance Sheet Arrangements
The Company is not party to any off-balance sheet transactions. The Company has no guarantees or obligations other than those which arise out of normal business operations.
Critical Accounting Policies and Significant Judgments and Estimates
The Company’s management’s discussion and analysis of its financial condition and results of operations is based on its financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States of America (“GAAP”). The preparation of these financial statements requires Apimeds Pharmaceuticals US, Inc. to make estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities as of the date of the balance sheet and the reported amounts of expenses during the reporting period. In accordance with GAAP, Apimeds Pharmaceuticals US, Inc. evaluates its estimates and judgments on an ongoing basis. The most significant estimates relate to convertible instruments. Apimeds Pharmaceuticals US, Inc. bases its estimates and assumptions on current facts, historical experiences, and various other factors that Apimeds Pharmaceuticals US, Inc. believes are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
The Company defines its critical accounting policies as those accounting principles that require it to make subjective estimates and judgments about matters that are uncertain and are likely to have a material impact on its financial condition and results of operations, as well as the specific manner in which the Company applies those principles. While its significant accounting policies are more fully described in Note 2 to its financial statements, the Company believes the following are the critical accounting policies used in the preparation of its financial statements that require significant estimates and judgments.
Convertible Instruments
The Company evaluates and accounts for conversion options embedded in convertible instruments in accordance with ASC 815 “Derivatives and Hedging Activities”.
The Company accounts for convertible instruments (when we have determined that the embedded conversion options should not be bifurcated from their host instruments) as follows: The Company records when necessary, discounts to convertible notes for the intrinsic value of conversion options embedded in debt instruments based upon the differences between the fair value of the underlying common stock at the commitment date of the note transaction and the effective conversion price embedded in the note. Debt discounts under these arrangements are amortized over the term of the related debt to their stated date of redemption.
31
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
As a smaller reporting company, we are not required to provide the information required by this Item.
Item 8. Financial Statements and Supplementary Data
The financial statements required pursuant to this item are included in Part IV, Item 15 of this Annual Report, beginning on page F-1.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
(a) Evaluation of Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as such term is defined under Rule 13a-15(e) promulgated under the Exchange Act, designed to ensure that information required to be disclosed in our reports filed pursuant to the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and our principal financial officer, as appropriate to allow timely decisions regarding required disclosures.
In designing and evaluating the disclosure controls and procedures, we recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and we were required to apply our judgment in evaluating the cost-benefit relationship of possible controls and procedures. We have carried out an evaluation as of December 31, 2024 under the supervision, and with the participation, of our management, including our Chief Executive Officer (who serves as our principal executive officer) and our Chief Financial Officer (who serves as our principal financial officer), of the effectiveness of the design and operation of our disclosure controls and procedures.
Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2024 in providing reasonable assurance of achieving the desired control objectives.
32
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting refers to the process designed by, or under the supervision of, our principal executive officer and principal financial officer, and effected by our Board, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that:
| (1) | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets |
| (2) | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and |
| (3) | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisitions, use or disposition of our assets that could have a material effect on the financial statements. |
Internal control over financial reporting has inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
We have conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2024, based on the framework established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO Framework). This assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of those controls. Based on that evaluation, as a result of the material weaknesses described below, management has concluded that our internal control over financial reporting was not effective as of December 31, 2024.
A material weakness in internal controls is a deficiency in internal control, or combination of control deficiencies, that adversely affects our ability to initiate, authorize, record, process, or report external financial data reliably in accordance with GAAP such that there is more than a remote likelihood that a material misstatement of our annual or interim financial statements that is more than inconsequential will not be prevented or detected. In the course of making our assessment of the effectiveness of internal controls over financial reporting, we identified material weaknesses in our internal control over financial reporting. Specifically, we do not have sufficiently documented procedures or control activities in place to support a reliable financial reporting process. This includes an absence of controls over the review and approval of journal entries, segregation of duties, reconciliations, and other fundamental accounting processes.
Based on our assessment under the criteria described above, we have concluded that our internal control over financial reporting was not effective as of December 31, 2024.
(b) Changes in Internal Control Over Financial Reporting
There were no changes in the Company’s internal controls over financial reporting that occurred during the year ended December 31, 2024 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting. The Company continues to review its disclosure controls and procedures, including its internal control over financial reporting, and may from time to time make changes aimed at enhancing their effectiveness and to ensure that the Company’s systems evolve with its business.
Item 9B. Other Information
None of our officers or directors, as defined
in Rule 16a-1(f) of the Exchange Act,
Item 9C. Disclosure Regarding Foreign Jurisdiction that Prevents Inspections
Not applicable.
33
PART III - OTHER INFORMATION
Item 10. Directors, Executive Officers and Corporate Governance
Information Regarding Directors and Executive Officers
The following table sets forth information regarding our executive officers and non-employee directors.
| Name | Age | Position | ||
| Dr. Christopher Kim, MD | 74 | Chairman and Chief Medical Officer | ||
| Erik Emerson | 54 | Chief Executive Officer and Director | ||
| Mark Corrao | 67 | Chief Financial Officer | ||
| Jakap Koo | 64 | Director | ||
| Dr. Bennett Weintraub, PhD. | 56 | Director | ||
| Hankil Yoon | 62 | Director | ||
| Carol O’Donnell | 67 | Director | ||
| Elona Kogan | 55 | Director |
Christopher Kim, MD. — Chairman and Chief Medical Officer
Dr. Christopher Kim has been our Chairman and Chief Medical Officer since our inception and served as our interim Chief Executive Officer from July 2022 to September 2023. Dr. Kim is the inventor and developer of Apitox and the founder of Apimeds Korea, where he has served as a director since its inception. Mr. Kim served as the Chief Executive Officer of Apimeds Korea from May 2003 to August 2011. Prior to founding Apimeds Korea, Dr. Kim lead with the support of Guju Pharmaceuticals, clinical trials for Apitoxin in Korea, which was approved by the Korea Food and Drug Administration in 2003 for relief of pain and inflammation for patients with Osteoarthritis. In 2005, he began focusing on the clinical development of Apitox in the United States, including the first of two-Phase III clinical studies for Osteoarthritis. Prior to his time with Apimeds Korea, Dr. Kim served as the President of the International Pain Institute of New Jersey from January 1983 to May 2003, a center for chronic pain and other disabling diseases that conducted clinical research and provided treatment. He served as a professor at Biomedical Center, CHA Graduate School of Medicine in Korea from March 2005 to February 2017. Dr. Kim is a licensed physician in New Jersey, New York and Korea and a Pain Medicine Specialist (American Board). Over the past twenty years, Dr. Kim has treated thousands of chronically disabled patients with autoimmune diseases, including MS. Dr. Kim received his medical degree from the School of Medicine, CN University in Korea.
We believe Dr. Kim’s extensive experience in pharmaceutical development and the biopharmaceutical industry, as well as his research and treatment of autoimmune diseases, and institutional knowledge of our product candidate, qualifies him to serve on our Board.
Erik Emerson — Chief Executive Officer
Erik Emerson has been our Chief Executive Officer since September 2023. Mr. Emerson is a 25-year veteran of the biopharmaceutical industry. He also serves as an advisor to Odyssey Neuropharma, Inc. where he has served since August 2022. In this role, Mr. Emerson leads business development and positioning efforts for a Phase II asset in evaluation for the treatment of mild traumatic brain injury (concussion). Mr. Emerson was the Chief Commercial Officer for Mezzion Pharmaceuticals, a Korean company establishing operations in the United States, for treatment of Single Ventricle Heart Disease post Fontan surgery, from February 2017 to January 2020. At Adhera Therapeutics, previously known as Marina Biotech, Mr. Emerson served as the Chief Commercial Officer and board member from February 2018 to November 2019. Prior to that Mr. Emerson served as the Executive Chairman and Chief Executive Officer of BioMarisLLC from July 2017 to November 2019. He also served as the President and Chief Executive Officer of Symplmed Pharmaceuticals & Technologies from July 2013 to May 2018. From May 2010 to July 2013, he served as the Senior Director of Commercial Development, Xoma Ltd. He was the director of marketing at Gilead Sciences from May 2007 to May 2010. Mr. Emerson has served as an advisory board member to NuGen Medical Devices since August 2022. Mr. Emerson began his career in sales, sales training and marketing with King Pharmaceuticals from September 2001 to May 2007. Mr. Emerson received a Bachelor’s in Arts in Political Science from the University of Oregon.
We believe Mr. Erikson’s extensive experience in the biopharmaceutical industry, as well as his prior executive-level experience at similarly situated companies, qualifies him to serve on our Board.
34
Mark Corrao — Chief Financial Officer
Mr. Mark Corrao has served as our Chief Financial Officer since October 2024. Mr. Corrao is currently serving as the chief financial officer for Ealixir, Inc. (OTC:EAXR), a publicly traded software company specializing in the management and protection of digital identities. He began serving in this role in January 2024. Previously, Mr. Corrao was the chief financial officer for Amesite, Inc. (OTC:AMST), a publicly traded software company from December 2021 through December 2022. Since February 2012, Mr. Corrao has served as the chief financial officer of Neuropathix, Inc., a private biopharmaceutical company. From June 2012 to July 2020 Mr. Corrao was a Managing Director of The CFO Squad LLC, where he is currently an advisor. From January 2017 to June 2021 Mr. Corrao was the chief financial officer for Generex Biotechnology Corp (OTC:GNBT) and its subsidiaries. From December 2018 to October 2021, Mr. Corrao was the chief financial officer for Brain Scientific, Inc., a medical device company. Mr. Corrao served as the chairman of the audit committee for Success Holdings Group International from January 2015 through December 2017. In February 2003, Mr. Corrao founded Strikeforce Technology, Inc. (OTC:SFOR), a publicly traded software development and services company and served as the chief financial officer until June 2010, and he remained a board member until August 2013. Prior to starting Strikeforce, Mr. Corrao was a director at Applied Digital Solutions from December 2000 through December 2001. Mr. Corrao was one of the founders and a Vice President at Advanced Communication Sciences from June 1997 through December 2000, when the company was sold. Mr. Corrao has spent numerous years in the public accounting arena specializing in certified auditing, SEC accounting, corporate taxation and financial planning. Mr. Corrao’s background also includes numerous years on Wall Street with Merrill Lynch, Spear Leeds & Kellogg and Greenfield Arbitrage Partners. While on Wall Street, Mr. Corrao was involved in several initial public offerings and has been a guiding influence in several startup companies. Mr. Corrao has a B.S. in Accounting from The City University of New York.
Jakap Koo — Director
Mr. Jakap Koo has served as a director since October 2023. Mr. Koo is also the Chief Executive Officer and President of both Apimeds Korea and its parent company, Inscobee Inc. (KRX: 006490), where he has served since March 2020. Before joining Apimeds Korea and Inscobee, from March 2015 to December 2019 he served as the Chief Executive Officer at Lotte Auto Lease Co. Ltd., where he grew company revenue through various financial services of car rental, installment payment, automobile leasing and investment banking to both B2B and B2C clients. Mr. Koo has spent more than 35 years mostly as C-level executives in various financial institutions and IT companies. His management and operational experiences cover banking, asset management, venture capital, private equity, and biotechnology companies. Mr. Koo has received his MBA from Stern School of New York University. He graduated from Seoul National University majoring in Law.
We believe Mr. Koo’s extensive financial knowledge qualifies him to serve on our Board.
Independent Directors:
Dr. Bennett Weintraub, PhD.
Dr. Weintraub has served as a director since October 2023. Dr. Weintraub currently serves as the President of inThought Research (“inThought”), a healthcare business intelligence consulting firm which he founded in 2009. inThought provides business development support, competitive intelligence monitoring, medical conference coverage, and other services both to professional investors and to pharma/ biotech companies. Dr. Weintraub has also served as the Chief Scientific Officer of inPhronesis since 2018.
After completing his training in immunology and biochemistry, Dr. Weintraub co-founded Biotech Tracker, an online tool for investors, where he served as a financial analyst from 2000 to 2008. From 2006 to 2008, Dr. Weintraub served as an analyst at Reuters Insight, providing analysis of drug development and trends in medicine to professional investors. Dr. Weintraub served as a licensed security analyst with Variant Research from 2005 to 2006.
35
From 1999 to 2000, Dr. Weintraub was senior scientific editor for the biology research journals Cell and Molecular Cell. Dr. Weintraub performed biochemistry and immunology research at Stanford University and at the John Curtin School of Medical Research in Canberra, Australia. He earned his doctorate in Biology from the University of California, San Diego, and a Bachelor of Science in Life Science from the Massachusetts Institute of Technology.
We believe Mr. Weintraub’s extensive science background qualifies him to serve on our Board.
Carol O’Donnell
Carol O’Donnell has served as a director since October 2023. Ms. O’Donnell is currently a Director and Member of the Audit Committee of Sono-Tek Corporation (NASDAQ: SOTK), where she has served since November 2018. Prior to that, she served as General Counsel to Boothbay Fund Management LLC, a registered investment adviser, from December 2019 through May 2021. Ms. O’Donnell joined Protégé Partners and MOV37, an industry leading firm investing in and seeding smaller and emerging hedge fund managers in April 2016 and has served as Chief Executive Officer since January 2018. Prior to joining Protégé Partners and MOV37, Ms. O’Donnell was the Director of Legal and Compliance with DARA Capital US, Inc., a Swiss-owned boutique registered investment advisory and wealth management firm from January 2013 to March 2016. She served as General Counsel and Chief Compliance Officer of each of the Permal Group and Framework Investment Group from June 2004 through February 2011 and from January 2002 to May 2004, respectively. She also served as a director of FSI Low Beta from 2012 to 2021. Ms. O’Donnell was named one of the Top 50 Women in Hedge Funds in September 2018 and is currently admitted to practice law in the State of Connecticut.
We believe Ms. O’Donnell’s extensive experience in the financial industry qualifies her to serve on our Board.
Hankil Yoon, PhD.
Hankil Yoon has served as a director since October 2023. Dr. Yoon has extensive experience in front-end business areas such as product strategy and planning, software technology and product development, mobile services, global partnership, sales, investment, and mergers and acquisitions and extensive knowledge of the entire software stack, ranging from firmware and OS, middleware. He is the owner of multiple patents on data mining and mobile technology.
Dr. Yoon was previously the Chief Executive Officer of Digital Domain Virtual Human, Inc. from January 2020 to November 2021 where he managed a global organization of developers throughout the United States, Canada and Taiwan using AI technology to implement best quality digital human at optimal speed using minimal amount of facial data and created partnerships with Google, Amazon, and Microsoft to implement “AI with a human face”. He served as the Executive Advisor to the Chief Executive Officer of Flipboard, Inc. from January 2019 to December 2020. Prior to that, Mr. Yoon served as the Senior Vice President at Samsung Electronics, from May 2005 to December 2018. He served as the Chairperson at the Tizen Association from January 2015 to December 2018. Mr. Yoon served as the Chief Technology Officer at Oracle Corporation, US, from August 2000 to May 2005. Dr. Yoon has BS in Computer Engineering, Seoul National University (1985) and an MBA (2017) in Global Management, an MS in Electrical & Computer Engineering, University of California at Irvine (1995), PhD, in Computer & Information Science & Engineering, University of Florida (2000).
We believe Mr. Yoon’s extensive experience in product strategy and planning, software technology and product development qualifies him to serve on our Board.
Elona Kogan
Elona Kogan has served as a director since October 2024. Beginning in August 2024, Ms. Kogan has served as the Chief Legal Officer of Terns Pharmaceutical, Inc. (Nasdaq: TERNS), a publicly traded biopharmaceutical company, Prior to joining us, from November 2020 through August 2024, Ms. Kogan served as the General Counsel and Chief Legal Officer of Seer Inc. (Nasdaq: SEER), a publicly traded life science company. From May 2018 through August 2021, Ms. Kogan served as a director of Cardax, Inc., a biotechnology company operating in the inflammatory health space. From March 2019 through August 2020, Ms. Kogan served as the General Counsel of Selecta Biosciences, Inc., a clinical-stage biotechnology company. Ms. Kogan is a graduate of Southwestern University School of Law. Ms. Kogan graduated from Columbia University, Barnard College, with a B.A. in Economics.
36
We believe Ms. Kogan’s extensive experience in biopharmaceutical and life science space, in addition to her experience serving as general counsel and chief legal officer of other publicly traded companies qualifies her to serve on our Board.
Family Relationships
There are no family relationships among any of our executive officers or directors.
Involvement in Certain Legal Proceedings
To the best of our knowledge, none of our executive officers or directors were involved in any legal proceedings described in Item 401(f) of Regulation S-K in the past ten years.
Compliance with Section 16(a) of the Exchange Act
Section 16(a) of the Securities Exchange Act of 1934, requires our directors, executive officers and persons who own more than 10% of our common stock to file with the SEC initial reports of ownership and reports of changes in ownership of common stock and other of our equity securities. During the year ended December 31, 2024, our officers, directors and 10% stockholders were not required to make filings pursuant to Section 16(a).
Code of Business Conduct and Ethics
In accordance with the information required by this Item 10 relating to the code of ethics required by Item 406 of Regulation S-K, the Company has a Code of Business Conduct and Ethics (the “Code”), which applies to its directors, officers (including its principal executive officer, the principal financial officer and principal accounting officer), and all other employees (collectively, the “Covered Persons” and each a “Covered Person”). The full text of the Code is available on the “Investors” section of the Company’s website. The Company intends to satisfy the SEC’s requirements regarding amendments to, or waivers from, the Code by posting such information on its website or by filing a Current Report on Form 8-K to disclose such information.
Procedures for Stockholders to Recommend Director Nominees
The Company’s bylaws (the “Bylaws”) were adopted on May 12, 2020. On February 7, 2025, the Company established the nominating and corporate governance committee of the Board and adopted the nominating and corporate governance committee’s written charter. Pursuant to the nominating and corporate governance committee’s charter, the committee may, if it deems appropriate, establish procedures to be followed by stockholders in submitting recommendations for Board candidates. Except as discussed in the foregoing sentences, there have been no material changes to the procedures by which security holders may recommend nominees to our Board.
Audit Committee Information
The Company’s Board has a standing audit committee. Our audit committee is chaired by Carol O’Donnell and its other members are Elona Kogan and Dr. Bennet Weintraub. Each member of the audit committee is financially literate. Carol O’Donnell qualifies as an “audit committee financial expert” as defined in applicable SEC rules.
Insider Trading Policy
The Company has an
37
Item 11. Executive Compensation
The following discussion contains forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs. The actual amount and form of compensation and the compensation policies and practices that we adopt in the future may differ materially from currently planned programs as summarized in this discussion.
As an “emerging growth company,” we have opted to comply with the executive compensation disclosure rules applicable to “smaller reporting companies,” as such term is defined in the rules promulgated under the Securities Act. Accordingly, we are required to provide a Summary Compensation Table, as well as limited narrative disclosures regarding executive compensation for our last two completed fiscal years and an Outstanding Equity Awards at Fiscal Year End Table for our last completed fiscal year. These reporting obligations extend only to “named executive officers.” Individuals we refer to as our “named executive officers” include (i) all individuals serving as our principal executive officer during the fiscal year ended December 31, 2024 and (ii) our two most highly compensated executive officers, as defined in Exchange Act Rule 3b-7, other than our principal executive officer, who were serving as executive officers at the end of the fiscal year ended December 31, 2024, whose salary and bonus for services rendered in all capacities exceeded $100,000 during the fiscal year ended December 31, 2024.
Our named executive officer for the year ended December 31, 2024, was our principal executive officer, Erik Emerson. No other executive officer of the Company received total compensation during the fiscal year ended December 31, 2024 in excess of $100,000, and thus disclosure is not required for any other person.
Summary Compensation Table
The following table sets forth information concerning the compensation of our named executive officer for the years ended December 31, 2024, and 2023.
| Name and Principal Position | Year | Salary ($) |
Bonus ($) |
Option Awards ($) |
Nonequity Incentive Plan Compensation ($) |
All Other Compensation ($) |
Total ($) |
|||||||||||||||||||
| Erik Emerson | 2024 | 300,000 | — | — | — | — | 300,000 | |||||||||||||||||||
| Chief Executive Officer | 2023 | 90,000 | — | — | — | — | 90,000 | |||||||||||||||||||
Narrative to Summary Compensation Table
Our executive compensation program is based on a pay for performance philosophy. Compensation for our Chief Executive Officer is composed primarily of the following main components: base salary, bonus, and equity incentives in the form of stock options. Like all full-time employees, our Chief Executive Officer is eligible to participate in our health and welfare benefit plans. As we transition from a private company to a publicly traded company, we intend to evaluate our compensation philosophy and compensation plans and arrangements as circumstances require.
Employment Agreement with Erik Emerson
The Company entered into an employment agreement with Erik Emerson on September 21, 2023 (the “Emerson Agreement”). Pursuant to the Emerson Employment Agreement, he will serve as the Company’s Chief Executive Officer and receive a yearly salary of $300,000. Mr. Emerson’s employment shall continue for one year from the date of execution and shall automatically renew for successive one year periods in the event of a closing on a public offering of the company during the initial term unless either party gives 30 days’ written notice of its intent not to renew the Emerson Agreement prior to the end of the then-current term.
If Mr. Emerson becomes disabled such that he is unable to perform his obligations hereunder, with or without reasonable accommodation, for a period of 180 days or more over a rolling consecutive twelve month period of time, or it is determined that Mr. Emerson is not able to perform the essential functions of his duties (incurs a “Disability”) the Company may terminate Mr. Emerson’s employment, unless otherwise required by law. In the event employment is terminated as a result of Mr. Emerson Disability, the Company shall have no further obligation to pay any unaccrued compensation or unaccrued benefits to Mr. Emerson for periods after the date of such termination.
38
Mr. Emerson may be terminated with or without Cause (as defined in the Emerson Agreement) upon thirty (30) days’ written notice to Mr. Emerson.
Mr. Emerson will not be eligible to receive an annual bonus at any time prior to the closing on a public offering (as defined in the Emerson Agreement) of the Company For all fiscal years following the closing on a public offering of the Company, including any fiscal year during which the closing on a public offering occurs, Mr. Emerson will be eligible to receive an annual bonus based on the achievement of goals for the Company’s and/or Mr. Emerson’s performance, as determined by the Board in its sole discretion.
Mr. Emerson is entitled to receive such other employee benefits and perquisites offered by the Company to any of the Company’s similarly-situated corporate employees, provided that the Company shall retain discretion to cancel, modify or amend such benefits provided to Mr. Emerson and similarly situated employees in its discretion.
Upon the closing on a public offering, Mr. Emerson shall receive an incentive stock option to purchase a number of shares of the Company’s common stock equal to 3% of the post-public offering capitalization of the Company (the “Equity Award”), of which 40% of the options shall vest upon grant and the remainder will vest in three equal installments on the annual anniversary of the date of grant. Mr. Emerson will agree not to sell any shares underlying the Equity Award, even if exercised, for a period of three years from the date of grant. Mr. Emerson will be eligible for future equity incentive awards in the discretion of the Board.
Mr. Emerson irrevocably assigns to the Company (or its designees), and agrees to hold in trust for the sole right and benefit of the Company, without any additional consideration, and to promptly make full written disclosure to the Company of, all of his right, title, and interest in and to any and all Inventions (as defined in the Emerson Agreement) that Mr. Emerson invents during his employment and for a period of one year following the termination of his employment with the Company.
There is customary confidentiality and non-solicitation clauses in Mr. Emerson’s agreement whereby he has agreed to keep all confidential information confidential and will not directly or indirectly solicit any of the Company’s employees or vendors after his employment with the Company ends.
While the Company employs Mr. Emerson, he agrees that he will not, without the Board’s prior written consent, directly or indirectly, provide services to any other person for which Mr. Emerson receives compensation, nor will he otherwise engage in activities that would conflict or interfere with his full and faithful performance of his duties as an employee of the Company.
Stock Option Award to Dr. Christopher Kim
On May 12, 2020 the Company granted Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer, a non-qualified stock option award to purchase 138,900 shares of the Company’s common stock at an exercise price of $11.28 per share. The option vested in three equal installments and vested fully on May 12, 2023. The options have a term of ten years from the date of grant and shall terminate at the expiration of that period, unless it is terminated at an earlier date pursuant to the provisions of the option grant agreement between the Company and Dr. Kim.
Consulting Agreement with Mark Corrao
On October 4, 2024, the Company entered into a consulting agreement with Mark Corrao (the “CFO Consulting Agreement”) to engage Mr. Corrao (the “Consultant”), to provide consulting services as the Company’s non-employee chief financial officer prior to the completion of the Company’s initial public offering. It is anticipated that following the completion of the Company’s initial public offering, the Consultant will become an employee of the Company on a full-time basis.
The Consultant has been duly appointed as the chief financial officer and principal financial and accounting officer of the Company and will remain as an executive officer of the Company during the term of the CFO Consulting Agreement. The Consultant will report directly to Erik Emerson, Chief Executive Officer and to any other party designated by Mr. Emerson in connection with the performance of the duties under the CFO Consulting Agreement and shall fulfill any other duties reasonably requested by the Company and agreed to by the Consultant.
39
The initial term of the CFO Consulting Agreement is one year. The CFO Consulting Agreement may only be extended thereafter by mutual agreement, unless earlier terminated. Either party may terminate the CFO Consulting Agreement at any time by providing thirty days’ written notice to the other party.
As compensation for the services rendered pursuant to the CFO Consulting Agreement, the Company shall pay Consultant a minimum $2,500 upon signing, and $2,500 per month for up to eight hours of services rendered per month, payable on the first business day of each month. Additional hours in excess of eight hours per month, if any, shall be billed at $250.00 per hour.
Outstanding Equity Awards at Fiscal-Year End 2024
There were no outstanding equity-based awards of the Company held by the named executive officer as of December 31, 2024.
Policies and Practices for Granting Certain Equity Awards
We do not schedule equity award grants in anticipation of the release of material nonpublic information, nor do we time the release of material nonpublic information based on equity grant dates.
Director Compensation Table
None of our directors received any form of compensation for the year ended December 31, 2024.
40
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters Securities
Authorized for Issuance under Share-Based Compensation Plans
Equity Compensation Plan Information
The following table sets forth, as of December 31, 2024, information regarding awards previously granted and outstanding, and securities authorized for future issuance, under the Company’s equity compensation plans.
| Plan Category | Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants or Rights |
Weighted-Average Exercise Price of Outstanding Options, Warrants or Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Outstanding Options, Warrants, or Rights) |
|||||||||
| Equity compensation plans approved by shareholders | — | — | 1,000,000 | |||||||||
| Equity compensation plans not approved by shareholders | 213,692 | 7.332 | — | |||||||||
| (1) | Represents shares available for grant under the Company’s Equity Incentive Plan (defined below) as of December 31, 2024. |
Apimeds Pharmaceuticals US, Inc. Equity Incentive Plan
On September 18, 2024, we adopted an equity incentive plan for our employees, the Apimeds Pharmaceuticals US, Inc. 2024 Equity Incentive Plan (the “Equity Incentive Plan”). The purposes of the Equity Incentive Plan are to provide additional incentives to selected employees, directors and independent contractors of, and consultants to, the Company or its affiliates, to strengthen their commitment, motivate them to faithfully and diligently perform their responsibilities and to attract and retain competent and dedicated persons who are essential to the success of our business and whose efforts will impact our long-term growth and profitability.
Awards
The Equity Incentive Plan allows the Company to make equity and equity-based incentive awards to officers, employees, directors, consultants, and advisors. The Board anticipates that providing such persons with a direct stake in the Company will assure a closer alignment of the interests of such individuals with those of the Company and its stockholders, thereby stimulating their efforts on the Company’s behalf and strengthening their desire to remain with the Company.
The Equity Incentive Plan provides for the grant of non-qualified stock options, incentive stock options, stock appreciation rights, restricted stock, restricted stock units, stock bonus awards, and performance compensation awards. All awards will be set forth in an award agreement which will detail all terms and conditions of the awards, including any applicable vesting and payment terms and post-termination exercise limitations.
A brief description of each award type follows.
| ● | Non-Qualified Stock Options means the right to purchase shares pursuant to terms and conditions that are not intended to be, or do not qualify as, an Incentive Stock Options; |
| ● | Incentive Stock Options means the right to purchase shares pursuant terms and conditions that are intended to qualify as, and that satisfy the requirements applicable to, an incentive equity option within the meaning of Code Section 422 of the United States Internal Revenue Code of 1986, as amended; |
| ● | Stock Appreciation Rights means a right, designated as an SAR, to receive the appreciation in the fair market value of shares; |
41
| ● | Restricted Stock means an award of shares subject to vesting conditions; |
| ● | Restricted Stock Units shall mean a right to receive shares or cash upon vesting; |
| ● | Stock Bonus Awards means unrestricted common stock, or other awards denominated in common stock, either alone or in tandem with other awards; and |
| ● | Performance Compensation Awards means an award granted to a participant that entitles the participant to delivery of shares or cash upon achievement of performance goals. |
1,000,000 shares of common stock have initially been reserved for the issuance of awards under the Equity Incentive Plan (the “Initial Limit”). The Initial Limit is subject to adjustment in the event of a reorganization, recapitalization, reclassification, stock split, stock dividend, reverse stock split or other similar change in the Company’s capitalization. The maximum aggregate number of shares of common stock of the Company that may be issued upon exercise of incentive stock options under the Equity Incentive Plan shall not exceed the Initial Limit, as adjusted. Shares underlying any awards under the Equity Incentive Plan that are forfeited, cancelled, held back upon exercise of an option or settlement of an award to cover the exercise price or tax withholding, satisfied without the issuance of stock or otherwise terminated (other than by exercise) will be added back to the shares available for issuance under the Equity Incentive Plan and, to the extent permitted under Section 422 of the Code and the regulations promulgated thereunder, the shares that may be issued as incentive stock options.
The Equity Incentive Plan is currently administered by a committee of at least two people as the Board may appoint to administer the Equity Incentive Plan or, if no such committee has been appointed by the Board, the Board, pursuant to the terms of the Equity Incentive Plan (the “Committee”). The plan administrator, which initially will be the Committee, has full power to select, from among the individuals eligible for awards, the individuals to whom awards will be granted, to make any combination of awards to participants, and to determine the specific terms and conditions of each award, subject to the provisions of the Equity Incentive Plan. The plan administrator may delegate to a committee consisting of one or more officers of the Company, the authority to awards to individuals who are not subject to the reporting and other provisions of Section 16 of the Exchange Act and not members of the delegated committee, subject to certain limitations and guidelines.
Persons eligible to participate in the Equity Incentive Plan will be officers, employees, non-employee directors, consultants, and advisors of the Company and its subsidiaries as selected from time to time by the plan administrator in its discretion. As of the date of this Annual Report, approximately 12 individuals are eligible to participate in the Equity Incentive Plan, which includes approximately two officers, no employees who are not officers, five non-employee directors, and five consultants/independent contractors.
Options
The Equity Incentive Plan permits the granting of both options to purchase common stock of the Company intended to qualify as incentive stock options under Section 422 of the Code and options that do not so qualify. Options granted under the Equity Incentive Plan will be non-qualified options if they fail to qualify as incentive stock options or exceed the annual limit on incentive stock options. Incentive stock options may only be granted to employees of the Company and its subsidiaries. Non-qualified options may be granted to any persons eligible to receive awards under the Equity Incentive Plan. The option exercise price of each option will be determined by the plan administrator but generally may not be less than 100% of the fair market value of the common stock of the Company on the date of grant or, in the case of an incentive stock option granted to a ten percent stockholder, 110% of such share’s fair market value. The term of each option will be fixed by the plan administrator and may not exceed ten years from the date of grant. The plan administrator will determine at what time or times each option may be exercised, including the ability to accelerate the vesting of such options.
Upon exercise of options, the option exercise price must be paid in full either in cash, by certified or bank check or other instrument acceptable to the plan administrator or by delivery (or attestation to the ownership) of shares of common stock of the Company that are beneficially owned by the optionee free of restrictions or were purchased in the open market. Subject to applicable law, the exercise price may also be delivered by a broker pursuant to irrevocable instructions to the broker from the optionee. In addition, the plan administrator may permit non-qualified options to be exercised using a “net exercise” arrangement that reduces the number of shares issued to the optionee by the largest whole number of shares with fair market value that does not exceed the aggregate exercise price.
42
Stock Appreciation Rights
The plan administrator may award stock appreciation rights subject to such conditions and restrictions as it may determine. Stock appreciation rights entitle the recipient to shares of common stock of the Company, or cash, equal to the value of the appreciation in the Company’s stock price over the exercise price. The exercise price generally may not be less than 100% of the fair market value of common stock of the Company on the date of grant. The term of each stock appreciation right will be fixed by the plan administrator and may not exceed ten years from the date of grant. The plan administrator will determine at what time or times each stock appreciation right may be exercised, including the ability to accelerate the vesting of such stock appreciation rights.
Restricted Stock and Restricted Stock Units
The plan administrator may award restricted shares of common stock of the Company and restricted stock units to participants subject to such conditions and restrictions as it may determine. These conditions and restrictions may include the achievement of certain performance goals and/or continued employment with the Company through a specified vesting period. The plan administrator may also grant shares of common stock of the Company that are free from any restrictions under the Equity Incentive Plan. Unrestricted stock may be granted to participants in recognition of past services or for other valid consideration and may be issued in lieu of cash compensation due to such participant. The plan administrator may grant dividend equivalent rights to participants that entitle the recipient to receive credits for dividends that would be paid if the recipient had held a specified number of shares of common stock of the Company.
Stock Bonus Awards
The plan administration may issue unrestricted common stock, or other awards denominated in common stock, under the Equity Incentive Plan to participants, either alone or in tandem with other awards, in such amounts as the plan administration shall from time to time in its sole discretion determine.
Performance Compensation Awards
The plan administrator may grant awards under the Equity Incentive Plan to participants, which may be cash-based, subject to the achievement of certain performance goals, including continued employment with the Company.
Other Material Features
The Equity Incentive Plan requires the plan administrator to make appropriate adjustments to the number of shares of common stock that are subject to the Equity Incentive Plan, to certain limits in the Equity Incentive Plan, and to any outstanding awards to reflect stock dividends, stock splits, extraordinary cash dividends and similar events.
Except as set forth in a stock award agreement issued under the Equity Incentive Plan, in the event of (i) a transfer of all or substantially all of the Company’s assets, (ii) a merger, consolidation or other capital reorganization or business combination transaction of the Company with or into another corporation, entity or person, or (iii) the consummation of a transaction, or series of related transactions, in which any person becomes the beneficial owner directly or indirectly, of more than 50% of Company’s then outstanding capital stock, each outstanding stock award (vested or unvested) will be treated as the plan administrator determines, which may include (a) Company’s continuation of such outstanding stock awards (if Company is the surviving corporation); (b) the assumption of such outstanding stock awards by the surviving corporation or its parent; (c) the substitution by the surviving corporation or its parent of new stock options or other equity awards for such stock awards; (d) the cancellation of such stock awards in exchange for a payment to the participants equal to the excess of (1) the fair market value of the shares subject to such stock awards as of the closing date of such corporate transaction over (2) the exercise price or purchase price paid or to be paid (if any) for the shares subject to the stock awards (which payment may be subject to the same conditions that apply to the consideration that will be paid to holders of shares in connection with the transaction, subject to applicable law); or (e) the opportunity for participants to exercise the stock options prior to the occurrence of the corporate transaction and the termination (for no consideration) upon the consummation of such corporate transaction of any stock options not exercised prior thereto.
The Equity Incentive Plan provides that a stock award may be subject to additional acceleration of vesting and exercisability upon or after a “Change in Control” (as defined in the Equity Incentive Plan) as may be provided in the award agreement for such stock award or as may be provided in any other written agreement between the Company or any affiliate and the participant, but in the absence of such provision, no such acceleration will occur.
43
Participants in the Equity Incentive Plan are responsible for the payment of any federal, state or local taxes that the Company or its subsidiaries are required by law to withhold upon the exercise of options or stock appreciation rights or vesting of other awards. The plan administrator may cause any tax withholding obligation of the Company or its subsidiaries to be satisfied, in whole or in part, by the applicable entity withholding from shares of common stock of the Company to be issued pursuant to an award shares with an aggregate fair market value that would satisfy the withholding amount due. The plan administrator may also require any tax withholding obligation of the Company or its subsidiaries to be satisfied, in whole or in part, by an arrangement whereby a certain number of shares issued pursuant to any award are immediately sold and proceeds from such sale are remitted to the Company or its subsidiaries in an amount that would satisfy the withholding amount due.
The Equity Incentive Plan generally does not allow for the transfer or assignment of awards, other than by will or by the laws of descent and distribution or pursuant to a domestic relations order; however, the plan administrator may permit the transfer of non-qualified stock options by gift to an immediate family member, to trusts for the benefit of family members, or to partnerships in which such family members are the only partners.
The plan administrator may amend or discontinue the Equity Incentive Plan and the plan administrator may amend or cancel outstanding awards for purposes of satisfying changes in law or any other lawful purpose, but no such action may materially and adversely affect rights under an award without the holder’s consent. Certain amendments to the Equity Incentive Plan will require the approval of the Company’s stockholders. Generally, without shareholder approval, (i) no amendment or modification of the Equity Incentive Plan may reduce the exercise price of any stock option or the strike price of any stock appreciation right, (ii) the plan administrator may not cancel any outstanding stock option or stock appreciation right where the fair market value of the common stock underlying such stock option or stock appreciation right is less than its exercise price and replace it with a new option or stock appreciation right, another award or cash and (iii) the plan administrator may not take any other action that is considered a “repricing” for purposes of the shareholder approval rules of the applicable securities exchange.
All awards granted under the Equity Incentive Plan will be subject to recoupment in accordance with any clawback policy that Company is required to adopt pursuant to the listing standards of any national securities exchange or association on which Company securities are listed or as is otherwise required by the U.S. Dodd-Frank Wall Street Reform and Consumer Protection Act or other applicable law. In addition, the Board may impose such other clawback, recovery or recoupment provisions in a stock award agreement as the Board determines necessary or appropriate.
No options or stock appreciation rights may be granted under the Equity Incentive Plan after the date that is ten years from the Equity Incentive Plan Effective Date. No awards under the Equity Incentive Plan have been made prior to the date of this Annual Report.
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth, as of April 15, 2025, certain information as to the Company’s common stock beneficially owned by persons known by the Company to own in excess of 5% of the outstanding shares of such stock. In addition, the table includes information regarding the shares of the Company’s common stock beneficially owned by (i) each named executive officer, (ii) each of the Company’s directors and (iii) the Company’s directors and executive officers as a group. Management knows of no person, except as listed below, who beneficially owned more than 5% of the outstanding shares of the Company’s common stock as of April 15, 2025. Except as otherwise indicated, the information provided in the following table was obtained from filings with the SEC and the Company pursuant to the Exchange Act. For purposes of the following table, in accordance with Rule 13d-3 under the Exchange Act, a person is deemed to be the beneficial owner of any shares of the Company’s common stock which he or she has or shares, directly or indirectly, voting or investment power, or which he or she has the right to acquire beneficial ownership of at any time within 60 days after April 15, 2025. As used herein, “voting power” is the power to vote, or direct the voting of, shares, and “investment power” includes the power to dispose of, or direct the disposition of, such shares. Unless otherwise noted, each beneficial owner has sole voting and sole investment power over the shares beneficially owned. Unless otherwise noted, the business address of each of the following entities or individuals is 65-1277 Ki Rd., Kamuela, Hawaii 96743.
44
| Name of Beneficial Owner | Number of Shares Beneficially Owned |
% of Common Stock | ||||||
| Directors and Named Executive Officers: | ||||||||
| Christopher Kim, MD(1) | 213,692 | 2.54 | % | |||||
| Erik Emerson | — | — | ||||||
| Mark Corrao | — | — | ||||||
| Bennett Weintraub, PhD | — | — | ||||||
| Hankil Yoon | — | — | ||||||
| Carol O’Donnell | — | — | ||||||
| Jakap Koo | 615,385 | 7.32 | % | |||||
| All directors and officers as a group (7 individuals) | 829,077 | 9.86 | % | |||||
| 5% or Greater Stockholders: | ||||||||
| Inscobee Inc.(2) | 5,908,783 | 70.28 | % | |||||
| Dominus IB, Inc.(3) | 800,000 | 9.52 | % | |||||
| Seed 1 ho(4) | 600,000 | 7.14 | % | |||||
| (1) | Represents 213,692 shares issuable pursuant to outstanding options, which are exercisable within 60 days of the date hereof. |
| (2) | Represents 1,596,760 shares or 18.99% held directly by Inscobee Inc. and 4,312,023 shares or 51.29% held through its wholly owned subsidiary, Apimeds Inc. Inscobee Inc. has voting and investment control over the shares held by Apimeds Inc. Millenium Holdings has voting and investment control with respect to the shares held by Inscobee Inc. Millenium Holdings is controlled by You In Soo, and as such, Mr. Yoo may be deemed to have beneficial ownership over the shares held by both Inscobee Inc. and Apimeds Inc. The business address for Inscobee Inc. is Room 613, Digital-ro 130, 6F, Geumcheon-gu, Seoul, 08580 Republic of Korea. The business address for Millenium Holdings is 107, Gasan Digital 2-ro, Geumcheon-gu, Seoul, Korea. Each of the parties named in this footnote disclaims any beneficial ownership of the reported shares other than to the extent of any pecuniary interest the party may have therein. |
| (3) | Dominus IB, Inc. is controlled by its Chief Executive Officer and largest shareholder, Park Kyoung Jin, who may be deemed to have voting and investment control with respect to the shares held by Dominus IB, Inc. The business address for Dominus IB, Inc. is 144, Dobong-ro, Gangbuk, Seoul, Republic of Korea. |
| (4) | Seed 1 ho is controlled by its Chief Executive Officer and largest shareholder, Son Hyoung Jin, who may be deemed to have voting and investment control with respect to the shares held by Seed 1 ho. The business address for Seed 1 ho is 116, Sindae-gil, Okcheon-myeon, Yangpyeong-gun, Gyeonggi-do, Republic of Korea. |
Changes in Control
Management of the Company knows of no arrangements, including any pledge by any person or securities of the Company, the operation of which may at a subsequent date result in a change in control of the registrant.
45
Item 13. Certain Relationships and Related Transactions, and Director Independence
Certain Relationships and Related Transactions
Other than as listed below, during 2024 and 2023, we were not a participant in any transaction or series of transactions in which the amount involved did exceed or may exceed the lesser of $120,000 or 1% of the average of our total assets at year-end for 2024 and 2023 in which any directors, director nominees, executive officers, greater than 5% beneficial owners and their respective immediate family members (each, a “Related Person”) had or will have a direct or indirect material interest, other than the compensation arrangements (including with respect to equity compensation) described in “Executive Compensation” beginning on page 38 and “Director Compensation” on page 40.
Business Agreement
On August 2, 2021, we entered into an agreement with Apimeds Korea, a principal stockholder of the Company (the “Business Agreement”). Pursuant to the Business Agreement, Apimeds Korea granted to the Company a sublicensable, royalty-bearing license to research, develop, manufacture and commercialize and sell Apitox in the United States. In exchange for this license, the Company will pay Apimeds Korea a perpetual royalty of 5% of the Company’s earnings before interest and taxes as determined consistent with GAAP, derived from the sale or license of Apitox, less any shipping, handling, and insurance charges, credits (arising from returns or other adjustments), discounts, rebates, or allowances of any kind (if any). The Business Agreement may be terminated by mutual written agreement by the parties and will automatically terminate upon the bankruptcy or dissolution of the Company.
Assignment Agreement
On October 12, 2021 we entered into an intellectual property assignment agreement (the “Assignment Agreement”) with Apimeds Korea and Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and founder of Apimeds Korea, effective as of May 12, 2021. Pursuant to the Assignment Agreement Dr. Kim transferred to Apimeds Korea all right, title, interest and good will in all of the intellectual property as it relates to Apitoxin, which will be marketed in the United States as Apitox (the “Assigned IP”).
Dr. Kim retained no right to use the Assigned IP. Additionally, the Assignment Agreement acknowledged that the Assigned IP was licensed to us to use via the Business Agreement, as described above.
Patent License Agreement
On October 12, 2021, we entered into a patent license agreement (the “Patent License Agreement”) Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and the founder of Apimeds Korea. During Dr. Kim’s engagement with Apimeds Korea, he contributed to the development of the intellectual property as it relates to Apitoxin. Pursuant to the Patent License Agreement, we were licensed certain patents. In consideration of its license under the Patent License Agreement, the Company paid Dr. Kim $1.00.
The patents expired in 2023 and, presently, the Company does not intend renew the expired patents or apply for any additional patents.
Business Establishment Agreement
On March 3, 2020, Apimeds Korea entered into a business establishment agreement with the Company pursuant to which Apimeds Korea agreed provide funding to us in the form of two tranches consisting of $500,000 each (for a total of $1,000,000). The first tranche was funded in March 2020 and the second tranche was funded in May 2020.
August 2021 Promissory Note
The Company issued to Apimeds Korea a convertible promissory note in the principal amount of $400,000, on August 30, 2021 (the “August 2021 Note”). The August 2021 Note is due and payable on the earlier of (i) August 30, 2026 or (ii) a sale of the Company (as defined in the August 2021 Note) (the “Maturity Date”). The August 2021 Note bears interest at an annual rate equal to the lesser of (i) 5% per annum, or (ii) the maximum rate permissible by law.
The Company may prepay the August 2021 Note at any time without penalty. If not previously paid by the Company, principal and accrued interest on the August 2021 Note will automatically convert into common stock (i) immediately prior to the closing of the Company’s firm commitment underwritten initial public offering resulting in at least $40,000,000 gross proceeds to the Company (a “Qualified IPO”), (ii) immediately prior to the closing of the Company’s initial listing of its common stock on an international exchange by means of an effective registration statement on Form S-1 that results in at least $40,000,000 of gross proceeds to the selling stockholders (a “Qualified Direct Listing”), or (iii) upon the consummation of the Company’s merger, consolidation, share exchange or other transaction with a publicly traded “special purpose acquisition company” resulting in a stock exchange listing (a “SPAC Transaction”). The number of shares of common stock shall be determined by dividing (x) the outstanding principal balance of the Apimeds Korea Note plus accrued but unpaid interest by (y) as applicable, (i) in case of a Qualified IPO, the per share price for which shares of common stock are initially offered in the Qualified IPO as reflected in the final prospectus, (b) in case of a Qualified Direct Listing, the fist closing price of the common stock on the first trading day, following the Qualified Direct Listing, and (c) in case of a SPAC Transaction, the price per share of the successor entity that is established in connection with such SPAC Transaction.
46
If there shall be any Event of Default (as defined below), the August 2021 Note shall accelerate and all principal and unpaid accrued interest shall become immediately due and payable, provided that the Company shall have 20 days from receipt of such notice to cure an Event of Default. The occurrence of any one or more of the following shall constitute an “Event of Default”: (a) the Company fails to pay timely all or any part of the principal amount or accrued interest due under the August 2021 Note, (b) the Company files any petition or action for relief under any bankruptcy, reorganization, insolvency or moratorium law or any other law for the relief of, or relating to, debtors, or makes any assignment for the benefit of creditors or takes any corporate action in furtherance of any of the foregoing, or (c) an involuntary petition is filed against the Company, or a custodian, receiver, trustee, assignee for the benefit of creditors (or other similar official) is appointed to take possession, custody or control of any property of the Company.
The terms of the August 2021 Note may only be amended with the written consent of both parties and my only be transferred upon its surrender to the Company for registration of transfer or accompanied by a duly executed written instrument of transfer in the form satisfactory to the Company.
On December 5, 2023, the Company and Apimeds Korea amended the August 2021 Note (the “August 2021 Note Amendment”) as follows: the maturity date was extended to the earlier of (i) December 31, 2026, or (ii) the consummation of an offering of our common stock (and other securities potentially) resulting in the listing for trading of our common stock on the NYSE American, the Nasdaq Capital Market, the Nasdaq Global Market, the Nasdaq Global Select Market or the New York Stock Exchange (or any successors to any of the foregoing) (“Qualified Offering”).
Additionally, the August 2021 Note Amendment provided for conversion of the note, including accrued and unpaid interest, at a conversion price of $2.60 per share as follows: (i) at the option of the holder, in its sole discretion, in whole or in part, and (ii) mandatorily simultaneous with the consummation of a Qualified Offering, in each case, into fully paid and nonassessable shares of common stock at the conversion price.
On June 12, 2024, Apimeds Korea assigned the August 2021 Note to Inscobee, Inc. a South Korean company, and the parent company of Apimeds Korea, (“Inscobee”).
If not converted earlier, upon the closing of a Qualified Offering, the August 2021 Note will automatically convert into approximately 179,283 shares of common stock.
March 2022 Promissory Note
The Company issued to Apimeds Korea, a promissory note in the principal amount of $160,000 on March 21, 2022 (the “March 2022 Note”). The March 2022 Note bears interest at a rate equal to 5% per annum (the “Interest Rate”). The March 2022 Note is due and payable on the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000, or (ii) July 15, 2022.
The Company may prepay the March 2022 Note at any time without penalty. If any payment due on the March 2022 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and Inscobee may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
On December 5, 2023, the Company and Apimeds Korea amended the March 2022 Note (the “March 2022 Note Amendment”) as follows: the maturity date was extended to the earlier of (i) December 31, 2026, or (ii) the consummation of a Qualified Offering.
Additionally, the March 2022 Note Amendment provided for conversion of the note, including accrued and unpaid interest, at a conversion price of $2.60 per share as follows: (i) at the option of the holder, in its sole discretion, in whole or in part, and (ii) mandatorily simultaneous with the consummation of a Qualified Offering, in each case, into fully paid and nonassessable shares of common stock at the conversion price.
On June 12, 2024, Apimeds Korea assigned the March 2022 Note to Inscobee.
If not converted earlier, upon the closing of a Qualified Offering, the March 2022 Note will automatically convert into approximately 70,002 shares of common stock.
47
June 2022 Promissory Note
On June 3, 2022, the Company issued to Inscobee, Inc. a South Korean company, and the parent company of Apimeds Korea, (“Inscobee”) a $100,000 promissory note (the “June 2022 Note”). Interest on the outstanding principal balance of the Second Loan accrues at a rate equal to 5% per annum, and interest on the outstanding principal balance of the First Loan shall accrue and be payable on the maturity date. The maturity date was the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000), and (ii) July 15, 2022.
On December 5, 2023, the Company and Inscobee amended the June 2022 Note (the “June 2022 Note Amendment”) as follows: the maturity date was extended to (i) December 31, 2026, or (ii) consummation of a Qualified Offering.
Additionally, the June 2022 Note Amendment provided for conversion of the note, including accrued and unpaid interest, at a conversion price of $2.60 per share as follows: (i) at the option of the holder, in its sole discretion, in whole or in part, and (ii) mandatorily simultaneous with the consummation of a Qualified Offering, in each case, into fully paid and nonassessable shares of common stock at the conversion price.
Upon the closing of a Qualified Offering, the June 2022 Note will automatically convert into approximately 43,361 shares of common stock.
On June 12, 2024, the Company and Inscobee amended the June 2022 Note to correct a scrivener’s error.
May 2024 Promissory Note
On May 20, 2024, the Company issued to Inscobee a $100,000 promissory note (the “May 2024 Note”). The May 2024 Note bears interest at a rate equal to 5% per annum (the “Interest Rate”). The May 2024 Note is due and payable on the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000, or (ii) May 19, 2025.
The Company may prepay the May 2024 Note at any time without penalty. If any payment due on the May 2024 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and Inscobee may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
August 2024 Note
On August 19, 2024, the Company issued to Inscobee a $150,000 principal amount promissory note (the “August 2024 Note”). The August 2024 Note bears interest at a rate equal to 5% per annum (the “Interest Rate”). The August 2024 Note is due and payable on the earlier of (i) the closing of an equity financing by the Company with gross proceeds to the Company of at least $3,000,000, or (ii) May 19, 2025. The outstanding principal and interest on the August 2024 Note will be repaid upon the closing of a Qualified Offering.
The Company may prepay the August 2024 Note at any time without penalty. If any payment due on the August 2024 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and Inscobee may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
March 2025 Promissory Note
On March 31, 2025, the Company received $250,000 in a promissory note (the “March 2025 Note”) agreement with Apimeds, Inc., one of its shareholders. The Promissory Notes bear interest at 5% per annum and mature on the earlier of (a) December 31, 2026 or (b) consummation of a Qualified Offering (the “Maturity Date”). “Qualified Offering” shall mean an offering of Common Stock (and other securities potentially) resulting in the listing for trading of the Common Stock on the NYSE American, the Nasdaq Capital Market, the Nasdaq Global Market, the Nasdaq Global Select Market or the New York Stock Exchange (or any successors to any of the foregoing).
The Company may prepay the March 2025 Note at any time without penalty. If any payment due on the March 2025 Note is not paid within five days after the amount becomes due, the payment shall be considered in default and the interest rate will increase by an additional 5% on the defaulted payment amount and may also, in its sole discretion, without notice or demand, declare the entire unpaid principal balance plus accrued interest due and payable immediately.
Cash Advance Loans
On October 5, 2022, November 10, 2022 and March 16, 2023, Dr. Christopher Kim, the Company’s Chairman and Chief Medical Officer and founder of Apimeds Korea, loaned the Company $9,900, $13,000 and $9,000 respectively. These loans carried no interest and did not have a maturity date. The loans were used for operating purposes. As of September 2023, all the loan amounts were repaid.
48
Policies and Procedures for Transaction with Related Persons
It is the responsibility of our audit committee to review and approval all related party transactions that would need to be disclosed pursuant to Item 404(a) of Regulation S-K (each a “Related Party Transaction”). The Board has adopted a related party transaction policy that makes up a part of the audit committee’s charter (the “Related Party Transactions Policy”). Pursuant to the Related Party Transactions Policy, each of the Company’s directors and executive officers shall promptly inform the chairperson of the audit committee of any potential Related Party Transactions. In addition, each such director and executive officer shall complete a questionnaire on an annual basis designed to elicit information about any potential Related Party Transactions. Any potential Related Party Transactions that are brought to the audit committee’s attention shall be analyzed by the audit committee, in consultation with outside counsel or members of management, as appropriate, to determine whether the transaction or relationship does, in fact, constitute a Related Party Transaction requiring compliance with the Related Party Transactions Policy. In determining whether to approve a Related Party Transaction, the audit committee shall consider, among other factors, the following factors to the extent relevant to the Related Party Transaction: (i) whether the terms of the Related Party Transaction are fair to the Company and on the same basis as would apply if the transaction did not involve a Related Party (as defined in the Related Party Transactions Policy); (ii) whether there are business reasons for the Company to enter into the Related Party Transaction; (iii) whether the Related Party Transaction would impair the independence of an outside director; (iv) whether the Related Party Transaction would present an improper conflict of interest for any director or executive officer of the Company, taking into account the size of the transaction, the overall financial position of the director, executive officer or Related Party, the direct or indirect nature of the director’s, executive officer’s or Related Party’s interest in the transaction and the ongoing nature of any proposed relationship, and any other factors the Committee deems relevant; and (v) any pre-existing contractual obligations. All of the transactions described in this section occurred prior to the adoption of this policy.
Director Independence
The Company’s Board has determined that Dr. Bennet Weintraub PhD, Dr. Hankil Yoon PhD, Carol O’Donnell and Elona Kogan, who together comprise a majority of the Board, are independent under applicable rules and regulations of the SEC. The Board made such independence determinations using the definition of independence set forth in the rules of the NYSE American based on a review of transactions and relationships between each director or any member of his or her immediate family, on the one hand, and the Company and its subsidiaries and affiliates, on the other hand, as well as transactions and relationships between each director or his affiliates, on the one hand, and members of the Company’s management or their affiliates, on the other hand.
Item 14. Principal Accountant Fees and Services
Kreit & Chiu CPA LLP served as the independent registered public accounting firm for the Company for 2024 and 2023. The following table sets forth the fees billed to the Company by Kreit & Chiu CPA LLP for 2024 and 2023.
| 2024 | 2023 | |||||||
| (in thousands) | ||||||||
| Audit Fees (1) | $ | 142,918 | $ | 74,950 | ||||
| Audit-Related Fees | - | - | ||||||
| All Other Fees | - | - | ||||||
| Total Fees | $ | 142,918 | $ | 74,950 | ||||
(1) Represents, for each year, fees for services related to the Company’s annual financial statement audit and quarterly reviews.
Under its charter, the Company’s audit committee must review and pre-approve both audit and permitted non-audit services provided by the Company’s independent registered public accounting firm and shall not engage the independent registered public accounting firm to perform any non-audit services prohibited by law or regulation. The independent registered public accounting firm’s retention to audit the Company’s financial statements, including the associated fee, is subject to approval each year by the audit committee. The audit committee does not regularly evaluate potential engagements of the independent registered public accounting firm and approve or reject such potential engagements. At each audit committee meeting, the audit committee receives updates on the services actually provided by the independent registered public accounting firm, and management may present additional services for pre-approval. The audit committee may delegate to the chairman of the audit committee the authority to evaluate and approve engagements on behalf of the audit committee in the event that a need arises for pre-approval between regular audit committee meetings. If the chairman so approves any such engagements, he will report that approval to the full audit committee at the next audit committee meeting. The audit committee was established on February 7, 2025, and therefore, the Company’s audit committee did not pre-approve all of the foregoing services.
49
PART IV
Item 15. Exhibit and Financial Statement Schedules
| (a) | Documents filed as part of this report |
| (1) | All financial statements |
| (2) | Financial Statement Schedules |
All financial statement schedules are omitted because they are either inapplicable or not required, or because the required information is included in the Financial Statements or notes thereto contained in this Annual Report on Form 10-K.
| (3) | Exhibits required by Item 601 of Regulation S-K |
50
51
| 21.1 | Subsidiaries of the Registrant (incorporated herein by reference to Exhibit 21.1 to our Registration Statement on Form S-1 filed on September 25, 2024). | |
| 31.1* | Certification of Chief Executive Officer Pursuant to Securities Exchange Act Rules 13a-14(a), as adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31.2* | Certification of Chief Financial Officer Pursuant to Securities Exchange Act Rules 13a-14(a), as adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32.1* | Certification of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350, as adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2* | Certification of Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 97.1* | Executive Compensation Recovery Policy. | |
| 101.INS | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |
| 101.LAB | Inline XBRL Taxonomy Extension Labels Linkbase Document. | |
| 101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |
| 104 | Cover Page Interactive Data File (Embedded within the Inline XBRL document and included in Exhibit). | |
| 101.INS | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document. |
| * | Filed or furnished herewith. |
Item 16. Form 10-K Summary
None.
52
INDEX TO FINANCIAL STATEMENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Shareholders
Apimeds Pharmaceuticals US, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Apimeds Pharmaceuticals US, Inc. as of December 31, 2024 and 2023, and the related statements of operations, changes in shareholders’ equity (deficit), and cash flows for each of the two years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Apimeds Pharmaceuticals US, Inc. as of December 31, 2024 and 2023, and the results of its operations and its cash flows for each of the two years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the entity will continue as a going concern. As discussed in Note 1 to the financial statements, the entity has suffered recurring losses from operations and has accumulated deficit that raise substantial doubt about its ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the entity’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to Apimeds Pharmaceuticals US, Inc. in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Apimeds Pharmaceuticals US, Inc. is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the entity's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/
We have served as Apimeds Pharmaceuticals US, Inc.’s auditor since 2023.
April 15, 2025
F-2
Apimeds Pharmaceuticals US, Inc.
Balance Sheets
| December 31, | December 31, | |||||||
| 2024 | 2023 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash | $ | $ | ||||||
| Prepaid expenses and other current assets | ||||||||
| Total current assets | ||||||||
| Total assets | $ | $ | ||||||
| Liabilities and shareholders’ equity (deficit) | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Accrued interest - related party | ||||||||
| Advance payable to related party | ||||||||
| Notes payable - related party | ||||||||
| Total current liabilities | ||||||||
| Convertible note - related party | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies (note 6) | ||||||||
| Shareholders’ equity (deficit): | ||||||||
| Preferred stock, par value $ |
||||||||
| Common stock, par value $ |
||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( |
) | ( |
) | ||||
| Total shareholders’ equity (deficit) | ( |
) | ||||||
| Total liabilities and shareholders’ equity (deficit) | $ | $ | ||||||
The accompanying notes are an integral part of these financial statements.
F-3
Apimeds Pharmaceuticals US, Inc.
Statements of Operations
| For the year ended December 31, |
||||||||
| 2024 | 2023 | |||||||
| Operating expenses: | ||||||||
| Research and development expenses | $ | $ | ||||||
| General and administrative expenses | ||||||||
| Loss from operations | ( |
) | ( |
) | ||||
| Other (expenses) income | ||||||||
| Interest income | ||||||||
| Interest expense | ( |
) | ( |
) | ||||
| Total other expense | ( |
) | ( |
) | ||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Weighted average shares outstanding | ||||||||
| Basic and diluted loss per share | $ | ( |
) | $ | ( |
) | ||
The accompanying notes are an integral part of these financial statements.
F-4
Apimeds Pharmaceuticals US, Inc.
Statement of Changes in Shareholders’ Equity (Deficit)
| Preferred Stock | Common Stock | Additional | ||||||||||||||||||||||||||
| Number of Shares | Amount | Number of Shares | Amount | Paid-in capital | Accumulated Deficit |
Total | ||||||||||||||||||||||
| Balance at December 31, 2022 | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
| Stock-based compensation expense | - | - | ||||||||||||||||||||||||||
| Issuance of shares to shareholders | ||||||||||||||||||||||||||||
| Embedded conversion feature of convertible notes | - | - | ||||||||||||||||||||||||||
| Net loss | - | - | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||||||
| Net loss | - | - | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-5
Apimeds Pharmaceuticals US, Inc.
Statements of Cash flows
| For the Years Ended December 31, |
||||||||
| 2024 | 2023 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense | ||||||||
| Accrued interest expense - related parties | ||||||||
| Accretion expense | ||||||||
| Changes in operating assets and liabilities | ||||||||
| Prepaid expenses and other current assets | ( |
) | ||||||
| Accounts payable and accrued expenses | ||||||||
| Net cash used in operating activities | ( |
) | ( |
) | ||||
| Cash flows from investing activities: | ||||||||
| Net cash provided by investing activities | ||||||||
| Cash flows from financing activities: | ||||||||
| Proceeds from notes payable - related parties | ||||||||
| Cash advances from related parties | ||||||||
| Cash advances paid to related parties | ( |
) | ||||||
| Issuance of shares for cash received | ||||||||
| Net cash provided by financing activities | ||||||||
| Net (decrease) increase in cash | ( |
) | ||||||
| Cash, beginning of year | ||||||||
| Cash, end of year | $ | $ | ||||||
The accompanying notes are an integral part of these financial statements.
F-6
Apimeds Pharmaceuticals US, Inc.
Notes to Financial Statements
1. DESCRIPTION OF BUSINESS
Business Description
Apimeds Pharmaceuticals US, Inc. (the “Company” or “Apimeds”) was formed as a corporation in May 2020 and was incorporated in the State of Delaware. On August 21, 2021, Apimeds Inc., the shareholder of the Company (“Apimeds Korea”), and Apimeds Pharmaceuticals US Inc. entered into the business agreement, under which the Company was designated to operate a pharmaceutical business which provides the biological drug named Apitox™ to clients in the biological drug commercial transaction area.
Apimeds is a clinical stage company that is in the process of developing Apitox™, a proprietary intradermally administered bee venom-based toxin which completed a positive Phase 3 trial for the treatment of pain associated with Osteoarthritis in 2018 and is now proceeding with FDA discussions on next steps in approval. In the future, the Company plans to investigate potential uses for Apitox™ for in treating multiple sclerosis (“MS”), and intends to conduct non-registered corporate sponsorship studies to identify appropriate MS patient populations. Apitox™ is currently marketed and sold by Apimeds Korea in South Korea (Republic of Korea) as “Apitoxin” for the treatment of osteoarthritis. Apimeds Inc. holds the majority of the Company’s outstanding common stock and is a subsidiary of Inscobee Inc. (“Inscobee”).
The success of the Company is dependent on obtaining the necessary regulatory approvals of its product candidates, marketing its products and achieving profitable operations. The continuation of the research and development activities and the commercialization of its products, if approved, are dependent on the Company’s ability to successfully complete these activities and to obtain additional financing through a combination of financing activities and operations. It is not possible to predict either the outcome of future research and development or commercialization programs, or the Company’s ability to fund these programs.
Going Concern
The Company has evaluated whether there are any
conditions and events, considered in the aggregate, that raise substantial doubt about its ability to continue as a going concern within
one year beyond the issuance date of these financial statements. As of December 31, 2024, the Company had accumulated losses amount to
$
The accompanying financial statements have been prepared assuming the Company will continue to operate as a going concern, which contemplates the realization of assets and settlement of liabilities in the normal course of business, and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from uncertainty related to its ability to continue as a going concern.
If the Company is unable to obtain sufficient financial resources, its business, financial condition and results of operations will be materially and adversely affected. This could affect future development and business activities and potential future clinical studies and/or other future ventures. There can be no assurance that the Company will be able to obtain the needed financing on acceptable terms or at all.
F-7
2. BASIS OF PRESENTATION AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The Company has prepared its financial statements in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASUs”) promulgated by the Financial Accounting Standards Board (“FASB”).
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make certain estimates, judgements and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Significant estimates and assumptions made in the accompanying financial statements include, but are not limited to, stock-based compensation and estimates that are related to convertible instruments. Actual results could differ from those estimates, and such differences could be material to the financial statements.
Fair Value Measurement
The fair value of the Company’s financial assets and liabilities reflects management’s estimate of amounts that the Company would have received in connection with the sale of the assets or paid in connection with the transfer of the liabilities in an orderly transaction between market participants at the measurement date. In connection with measuring the fair value of its assets and liabilities, the Company seeks to maximize the use of observable inputs (market data obtained from independent sources) and to minimize the use of unobservable inputs (internal assumptions about how market participants would price assets and liabilities). The following fair value hierarchy is used to classify assets and liabilities based on the observable inputs and unobservable inputs used in order to value the assets and liabilities:
Level 1 — Quoted prices in active markets for identical assets or liabilities. An active market for an asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis.
Level 2 — Observable inputs other than Level 1 inputs. Examples of Level 2 inputs include quoted prices in active markets for similar assets or liabilities and quoted prices for identical assets or liabilities in markets that are not active.
Level 3 — Unobservable inputs based on the Company’s assessment of the assumptions that market participants would use in pricing the asset or liability.
In some circumstances, the inputs used to measure fair value might be categorized within different levels of the fair value hierarchy. In those instances, the fair value measurement is categorized in its entirety in the fair value hierarchy based on the lowest level input that is significant to the fair value measurement.
Common Stock Reverse Stock Split
On February 7,2025, the Board
approved and implemented a reverse stock split ratio of 1-for-2.6, which provided that every
Concentrations of Credit Risk
Financial instruments that potentially subject
the Company to concentration of credit risk consist of cash accounts in financial institutions which, at times, may exceed the federal
depository insurance corporation limit of $
F-8
Segment Information
Operating segments are identified as components
of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker
(“CODM”), or decision-making group, in making decisions on how to allocate resources and assess performance. The Company has
Cash
The Company considers all highly liquid investments with an original maturity of three months or less at the date of purchase to be cash equivalents. As of December 31, 2024 and 2023, the Company had no cash equivalents.
Accrued Expenses
Accrued expenses consist of accrued interest for the convertible and promissory notes held with related parties, monies owed to vendors, as well as others, such as the taxing authority and employees.
As December 31, 2024, and 2023, the accounts payable and accrued expenses balance consists of the following:
| As of December 31, | ||||||||
| 2024 | 2023 | |||||||
| Professional fees payable | $ | $ | ||||||
| Accrued compensation | ||||||||
| $ | $ | |||||||
Convertible Instruments
The Company evaluates and accounts for conversion options embedded in convertible instruments in accordance with ASC 815 “Derivatives and Hedging Activities”.
Applicable U.S. GAAP requires companies to bifurcate conversion options from their host instruments and account for them as free-standing derivative financial instruments according to certain criteria. The criteria include circumstances in which (a) the economic characteristics and risks of the embedded derivative instrument are not clearly and closely related to the economic characteristics and risks of the host contract, (b) the hybrid instrument that embodies both the embedded derivative instrument and the host contract is not re-measured at fair value under other U.S. GAAP with changes in fair value reported in earnings as they occur and (c) a separate instrument with the same terms as the embedded derivative instrument would be considered a derivative instrument.
The Company accounts for convertible instruments (when we have determined that the embedded conversion options should not be bifurcated from their host instruments) as follows: The Company records when necessary, discounts to convertible notes for the intrinsic value of conversion options embedded in debt instruments based upon the differences between the fair value of the underlying common stock at the commitment date of the note transaction and the effective conversion price embedded in the note. Debt discounts under these arrangements are accreted over the term of the related debt to their stated date of redemption.
If a security or instrument becomes convertible only upon the occurrence of a future event outside the control of the Company, or, is convertible from inception, but contains conversion terms that change upon the occurrence of a future event, then any contingent beneficial conversion feature is measured and recognized when the triggering event occurs and contingency has been resolved.
F-9
Patent Costs
All patent-related costs incurred in connection with filing and prosecuting patent applications are expensed as incurred due to the uncertainty about the recovery of the expenditure. Amounts incurred are classified as general and administrative expenses in the accompanying statements of operations.
Leases
The Company accounts for a contract as a lease when it has the right to direct the use of the asset for a period of time while obtaining substantially all of the asset’s economic benefits. The Company determines the initial classification and measurement of its right-of-use assets (“ROU”) and lease liabilities at the lease commencement date and thereafter if modified. ROU assets and liabilities are to be represented on the balance sheet at the present value of future minimum lease payments to be made over the lease term. The Company has elected as an accounting policy not to apply the recognition requirements in ASC 2016-02, Leases (“ASC 842”) to short-term leases. Short-term leases are leases that have a term of 12 months or less and do not include an option to purchase the underlying asset that the Company is reasonably certain to exercise. The Company recognizes the lease payments for short-term leases on a straight-line basis over the lease term. As of December 31, 2024 and 2023, the Company did not have leases that qualified as ROU assets.
Related Parties
The Company follows ASC 850, “Related Party Disclosures” for the identification of related parties and disclosure of related party transactions.
General and Administrative
General and administrative expenses consist primarily of management personnel costs, professional service fees, and other general overhead and facility costs, including rent and insurance, which relate to the Company’s general and administrative functions.
Research and Development
Research and development expenses consist primarily of consulting, regulatory and manufacturing related costs, third-party license fees and external costs of vendors engaged to conduct preclinical development activities. These costs are expensed as incurred and non-refundable prepayments for goods or services that will be used or rendered for future research and development activities are deferred and capitalized in prepaid expenses and other current assets.
The Company enters into arrangements with contract research organizations in connection with pre-clinical and clinical trials. Such arrangements often provide for payment prior to commencing the project or based upon predetermined milestones throughout the period during which services are expected to be performed. As part of the process of preparing the Company’s financial statements, management is required to estimate prepaid and accrued clinical trial expenses. The date on which services commence, the level of services performed on or before a given date, and the cost of such services are often determined based on subjective judgments informed by the facts and circumstances known to management from the terms of the contract and the Company’s ongoing monitoring of service performance. The Company makes these judgments based upon the facts and circumstances known to management based on the terms of the contract and the Company’s ongoing monitoring of service performance.
In line with the guidance suggested under ASC 450, Contingencies and ASC 730, Research and Development, all research and development costs will be expensed as incurred. Development and regulatory milestone payments are accounted for by estimating the probability of milestone achievement.
Stock Based Compensation
The Company accounts for share-based compensation in accordance with the fair value recognition provision of FASB ASC 718, Compensation – Stock Compensation (“ASC 718”), which prescribes accounting and reporting standards for all share-based payment transactions in which employee services are acquired. Transactions include incurring liabilities, or issuing or offering to issue shares, options, and other equity instruments such as employee stock ownership plans and stock appreciation rights. Share-based payments to employees, including grants of employee stock options, are recognized as compensation expense in the financial statements based on the estimated grant date fair values. That expense is recognized over the period during which an employee is required to provide services in exchange for the award, known as the requisite service period (usually the vesting period). The Company accounts for forfeitures as they occur. The Company classifies share-based compensation expense in its statements of operations in the same manner in which the award recipient’s cash compensation costs are classified.
F-10
Given the absence of an active market for the Company’s equity, the Company and the board of directors were required to estimate the fair value of the Company’s common stock and equity awards at the time of each grant. The Company and the board of directors determined the estimated fair value of the Company’s equity instruments based on a number of factors, including external market conditions affecting the pharmaceutical industry sector. The Company and the board of directors utilized various valuation methodologies in accordance with the framework of the American Institute of Certified Public Accountants’ Technical Practice Aid, Valuation of Privately Held Company Equity Securities Issued as Compensation, to estimate the fair value of its equity instrument. Each valuation methodology includes estimates and assumptions that require the Company’s judgment.
Income Taxes
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences attributable to differences between carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax reporting purposes and for operating loss and tax credit carryforwards. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes.
The Company’s deferred tax assets and liabilities are measured using enacted tax rates expected to apply in the years in which these temporary differences are expected to be recovered or settled. A valuation allowance is recorded to reduce deferred tax assets if it is determined that it is more likely than not that all or a portion of the deferred tax asset will not be realized. The Company considers many factors when assessing the likelihood of future realization of deferred tax assets, including recent earnings results, expectations of future taxable income, carryforward periods available and other relevant factors. The Company records changes in the required valuation allowance in the period that the determination is made.
The Company assesses its income tax position and
records tax benefits for all years subject to examination based upon management’s evaluation of the facts, circumstances and information
available as of the reporting date. For those tax positions where it is more likely than not that a tax benefit will be sustained, the
Company records the largest amount of tax benefit with a greater than
Basic and Diluted Loss per share
Basic loss per share data for each period presented is computed using the weighted average number of shares of common stock outstanding during each such period. Diluted net loss per share is computed by giving effect to all potential shares of common stock to the extent they are dilutive.
F-11
The following table sets forth the number of potential shares of common stock that have been excluded from basic net loss per share because their effect was anti-dilutive:
| As of December 31, | ||||||||
| 2024 | 2023 | |||||||
| Employee stock options | ||||||||
| Convertible notes and interest | ||||||||
Emerging Growth Company
The Company intends to elect as an Emerging Growth Company, as defined in Section 2(a) of the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”). Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act, until such time as those standards apply to private companies. The Company has elected to use this extended transition period for complying with new or revised accounting standards that have different effective dates for public and private companies until the earlier of the date that it (i) is no longer an emerging growth company or (ii) affirmatively and irrevocably opts out of the extended transition period provided in the JOBS Act. As a result, these financial statements may not be comparable to companies that comply with the new or revised accounting pronouncements as of public company effective dates.
Prior period reclassifications
We have reclassified certain amounts in prior
periods to conform with current presentation. Accrued interest – related party in the amount of $
Recently Issued Accounting Pronouncements
The Company considers the applicability and impact of all Accounting Standard Updates. ASUs not discussed in these financial statements were assessed and determined to be either not applicable or are expected to have minimal impact on the financial statements.
In November 2023, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2023-07 - Segment Reporting (ASC 280): Improvements to Reportable Segment Disclosures, which enables investors to better understand an entity’s overall performance and assess potential future cash flows through improved reportable segment disclosure requirements. The amendments enhance disclosures about significant segment expenses, clarify circumstances in which an entity can disclose multiple segment measures of profit or loss, provide new segment disclosure requirements for entities with a single reportable segment, and contain other disclosure requirements. ASU 2023-07 is effective for annual periods beginning after December 15, 2023. The Company adopted ASU No. 2023-07 on December 31, 2024. The adoption of the standard did not result in any significant disclosure changes in the Notes to the Financial Statements.
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes – Improvements to Income Tax Disclosures (Topic 740). The amendments require that public business entities on an annual basis disclose specific categories in the rate reconciliation and provide additional information for reconciling items that meet a quantitative threshold. The amendments also require that all entities disclose on an annual basis the income taxes paid disaggregated by jurisdiction. The amendments eliminate the requirement for all entities to disclose the nature and estimate of the range of the reasonably possible change in the unrecognized tax benefits balance in the next 12 months or make a statement that an estimate of the range cannot be made. The amendments are effective for fiscal years beginning after December 15, 2024. The amendments should be applied on a prospective basis, although early adoption is permitted. The Company is currently evaluating the potential impact adopting ASU 2023-09 will have on the Company’s financial statements and related disclosures.
In November 2024, the FASB issued Accounting Standards Update No. 2024-03, Disaggregation of Income Statement Expenses. This guidance will require additional disclosures and disaggregation of certain costs and expenses presented on the face of the income statement. The amendments are effective for annual reporting periods beginning after December 15, 2026 and interim reporting period beginning after December 15, 2027 with early adoption permitted. The Company is currently evaluating the impact of this new guidance to our financial statements.
F-12
3. LICENSE AGREEMENTS
On August 2, 2021, the Company entered into a
business agreement with Apimeds Korea. Under the agreement, the Company received the right to continue any clinical trial and acquire
the permits and approval necessary from the U.S. Food and Drug Administration. The Company will pay Apimeds Korea a royalty of
On October 12, 2021, the Company entered into an exclusive patent license agreement with Apimeds Korea, a shareholder of the Company. Under the agreement, the Company was granted the exclusive right and license under the licensed patents to make and sell the licensed products in the United States of America.
The agreement shall commence on the effective
date and shall remain in force for each licensed product on a licensed-product-by-licensed-product basis for rights and obligations concerning
the licensed patent, until the expiration of the last to expire valid claim of a licensed patent. The total consideration exchanged for
the exclusive license agreement was $
4. DEBT
2022 Convertible notes (amended from notes payable) – related parties
On March 21, 2022, the Company entered into a
promissory note agreement in the amount of $
On December 5, 2023, the Company amended their
promissory notes to be convertible and extended the maturity date of the convertible notes with the related parties to be the earlier
of (i) December 31, 2026 or (ii) consummation of a qualified offering. The notes are convertible at a price of $
As of December 31, 2024 and 2023, there was accrued
interest in connection to the 2022 Convertible Notes of $
F-13
As of December
31, 2024 and 2023, the outstanding balance on the 2022 Convertible notes agreement, net of the unamortized debt discounts of $
2021 Convertible note – related party
On August 30, 2021, the Company received $
On December 5, 2023, the Company amended their
convertible note to be convertible at $
As
December 31, 2024 and 2023, there was accrued interest in connection with the 2021 Convertible Note of $
As of December 31,
2024 and 2023, the outstanding balance on the 2021 Convertible Note, net of the unamortized debt discounts of $
2024 Promissory Notes – Related Parties
On May
20, 2024, the Company received $
As of December
31, 2024, there was accrued interest in connection with the 2024 Promissory Notes of $
2024 Short Term Borrowing
On July 19, 2024, the Company entered into a non-interest-bearing
loan agreement with a private lender for $
F-14
5. ADVANCE PAYABLE – RELATED PARTY
As of December 31, 2024, the Company received
$
In March 2023, the Company received $
These advance payables carry no interest and do not have a maturity date. The cash proceeds from these advance payables were used for operating purposes.
6. COMMITMENTS AND CONTINGENCIES
Legal
Periodically, the Company reviews the status of any significant matters that exist and assesses its potential financial exposure. If the potential loss from any claim or legal claim is considered probable and the amount can be estimated, the Company accrues a liability for the estimated loss. Legal proceedings are subject to uncertainties, and the outcomes are difficult to predict. Because of such uncertainties, accruals are based on the best information available at the time. As additional information becomes available, the Company reassesses the potential liability related to pending claims and litigation. As of December 31, 2024 and 2023, there are no pending claims or litigation that are expected to materially affect the Company’s results going forward.
Executive employee agreement
On September 21, 2023, the Company signed an executive
employee agreement with the CEO of the Company. Under the executive employee agreement terms, if the Company closes on a public offering,
the CEO will be eligible to receive an incentive stock option to purchase a number of shares of the Company’s common stock equal
to
7. SHAREHOLDERS’ DEFICIT
Common Stock
As of December 31, 2024 and 2023, the Company
had
Preferred Stock
On December 5, 2023, the Company authorized
Activity during the period ended December 31, 2023
On September 7, 2023, the Company issued
On December 5, 2023, the Company established a preferred
stock class by authorizing
On December 6, 2023, the Company issued
F-15
8. STOCK-BASED COMPENSATION
Stock Options
On September 18, 2024, the Company adopted an equity incentive plan
for its employees, the Apimeds Pharmaceuticals US, Inc. 2024 Equity Incentive Plan (the “2024 Equity Incentive Plan”).
On May 12, 2020, the Company granted one of its
executive officers a total of
The following is a summary of stock options issued and outstanding as of December 31, 2024 and 2023:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Life (in years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding as of December 31, 2023 | $ | |||||||||||||||
| Granted | — | — | ||||||||||||||
| Exercised | — | — | ||||||||||||||
| Forfeited | — | — | ||||||||||||||
| Outstanding as of December 31, 2024 | $ | — | ||||||||||||||
| Exercisable as of December 31, 2024 | $ | |||||||||||||||
During the years ended December 31, 2024 and 2023,
there was $
The options were valued utilizing the Black-Scholes
options pricing model with the following inputs:
As of December 31, 2024, there were no remaining unrecognized compensation costs related to unvested options.
9. INCOME TAXES
There were no income tax expenses reflected in the results of operations for the years ended December 31, 2024 and 2023.
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Net loss per book | $ | ( |
) | $ | ( |
) | ||
| Federal statutory income tax rate ( |
( |
) | ( |
) | ||||
| State income tax, net of federal benefit | ( |
) | ( |
) | ||||
| State rate change | ||||||||
| Permanent item | ||||||||
| Prior period adjustment | ( |
) | ||||||
| Change in valuation allowance | ||||||||
| Income tax | $ | $ | ||||||
F-16
The tax effects of temporary differences which give rise to deferred tax assets (liabilities) are summarized as follows:
| Year Ended December 31, | ||||||||
| 2024 | 2023 | |||||||
| Net operating loss carry forwards | $ | $ | ||||||
| Stock-based compensation | ||||||||
Accrued compensation |
||||||||
| Capitalized research and development | ||||||||
| Intangible assets | ( |
) | ( |
) | ||||
| Total deferred tax assets | ||||||||
| Valuation allowance | ( |
) | ( |
) | ||||
| Net deferred tax assets | $ | $ | ||||||
The Company had cumulative federal net operating losses of approximately
$
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Deferred tax assets consist primarily of the tax effect of NOL carry-forwards. The Company has provided a full valuation allowance on the deferred tax assets because of the uncertainty regarding its realizability.
The Company’s policy is to record interest and penalties associated with unrecognized tax benefits as additional income taxes in the statement of operations. As of December 31, 2024, the Company had no unrecognized tax benefits. There were no changes in the Company’s unrecognized tax benefits during the years ended December 31, 2024 and 2023. The Company did not recognize any interest or penalties during the 2024 fiscal year related to unrecognized tax benefits.
10. SUBSEQUENT EVENTS
The Company evaluated subsequent events through the issuance date of the financial statements and determined that there have been no subsequent events except those mentioned throughout the footnotes that would require recognition in the financial statements or disclosure in the notes to the financial statements.
Subsequent to the year ended December 31, 2024,
the Company received an additional $
March 2025 Promissory Note
On March
31, 2025, the Company received $
The Company may prepay the March 2025 Note at any time without penalty.
If any payment due on the March 2025 Note is not paid within five days after the amount becomes due, the payment shall be considered in
default and the interest rate will increase by an additional
F-17
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| APIMEDS PharmaCEUTICALS US, Inc. | ||
| Date: April 15, 2025 | /s/ Erik C. Emerson | |
| Name: | Erik C. Emerson | |
| Title: | Chief Executive Officer | |
| (Principal Executive Officer)t | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
| SIGNATURE | TITLE | DATE | ||
| /s/ Erik Emerson | Chief Executive Officer and Director | April 15, 2025 |
||
| Erik Emerson | (Principal Executive Officer) | |||
| /s/ Mark Corrao | Chief Financial Officer | April 15, 2025 | ||
| Mark Corrao | (Principal Financial Officer and Principal Accounting Officer) | |||
| /s/ Dr. Christopher Kim | Chairman of the Board and Chief Medical Officer | April 15, 2025 | ||
| Dr. Christopher Kim | ||||
| /s/ Jakap Koo | Director | April 15, 2025 | ||
| Jakap Koo | ||||
| /s/ Bennett Weintraub, PhD | Director | April 15, 2025 | ||
| Bennett Weintraub, PhD | ||||
| /s/ Hankil Yoon | Director | April 15, 2025 | ||
| Hankil Yoon | ||||
| /s/ Carol O’Donnell | Director | April 15, 2025 | ||
| Carol O’Donnell | ||||
| /s/ Elona Kogan | Director | April 15, 2025 | ||
| Elona Kogan |
53